2025 AIChE Annual Meeting
(271c) Deep Learning Founded Alternative to High Throughput Screening for Noncanonical Amino Acid Incorporation
Authors
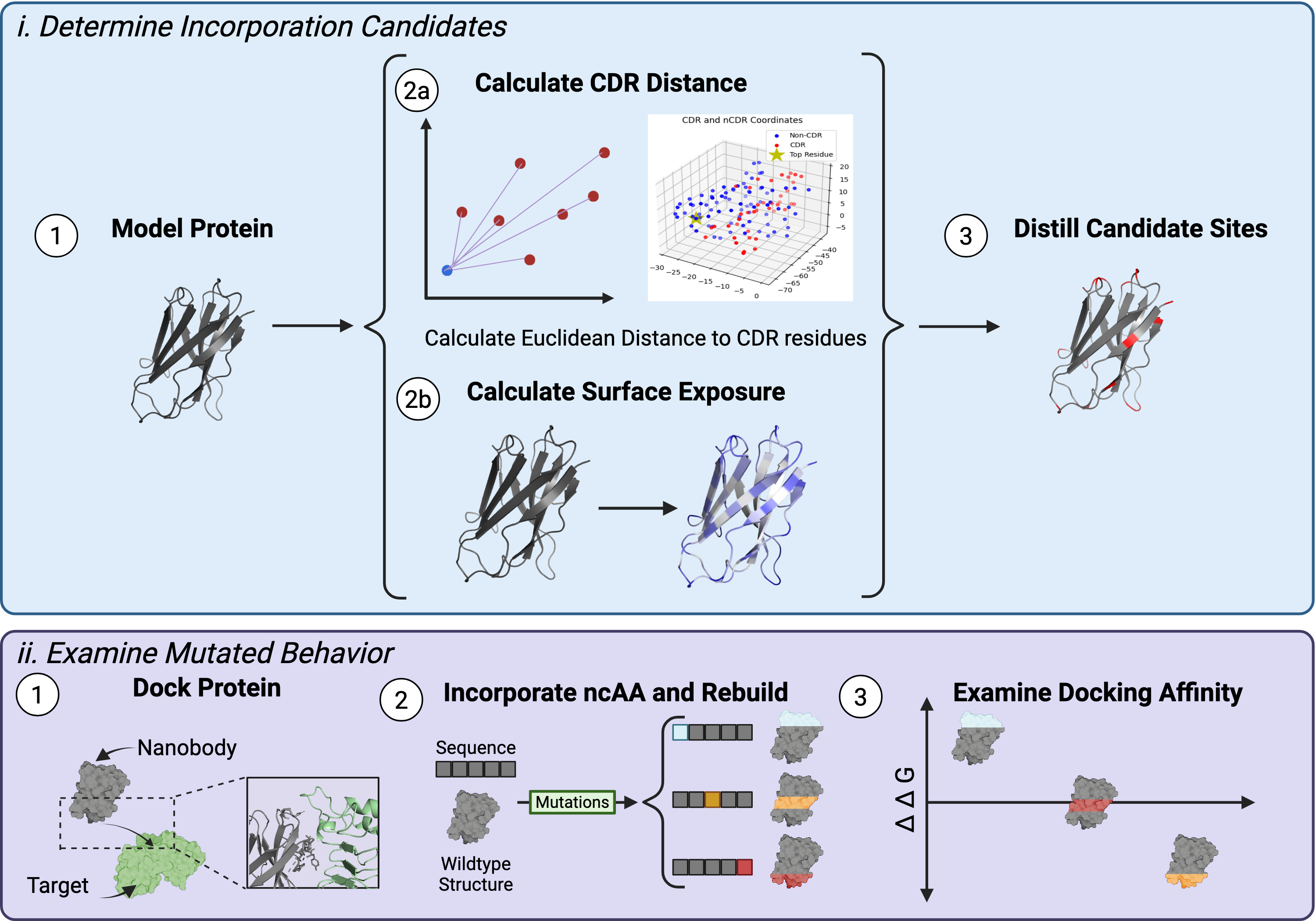
We seek to algorithmically discover these guidelines, creating a workflow in silico in which conjugation sites can be rationally selected based off a variety of parameters. By this introduction of computational methods, we quickly screen and select promising conjugation site candidates for more informed mutagenesis rounds. The project explores (1) the selection of the decision space from which the model will train off and (2) the model architecture itself. Due to the size of proteins and the data-dependent nature of deep learning models, we increased the density of our decision space through rationally selecting which residues to train off and the number of NCAAs explored (200+ unique NCAAs). Amino acid surface exposure and distance from the binding complementary determining region were screened and mutated independently to one another, with combination of screens tested afterwards. In the end, 60 total sites (20 from each screening process) were selected and mutated using the NCAA database. After designing our decision space, we moved into training our model. The model views proteins through the lens of a language, where each amino acid functions as a word to build out the sequence. Through understanding this protein language, the model can identify sensical locations to incorporate non-canonicals based on its characteristics. We then characterize the function of resultant nanobodies through examination of cell viability and target receptor binding affinity in vitro using relevant disease model cells. Upon collection of data, we plan to revisit the original computational model, leveraging statistical analysis to identify key features that have the highest correlation to both the binding interaction and intercellular activity, with the end goal of creating a predictive model accounting for necessary parameters. Through this, we can not only improve the accessibility of engineering NCAA containing proteins but also distill a deeper understanding of a protein's resiliency to structural perturbations.