2024 AIChE Annual Meeting
(175bj) Causal Discovery Algorithms Identify DNA Methylation Sites That Differentiate COPD Progression
Author
Methods. DNA methylation data was obtained from COPDGene, a multicenter observational study aimed at identifying COPD subtypes based on clinical measurements, chest CT phenotypes, and genetic variations. DNA was extracted from a total of 734 whole blood samples and processed with Illumina HumanMethylation 450K arrays to determine methylation status across 450,000 sites. For computational feasibility, non-negative matrix factorization (NMF) was used for dimensionality reduction. Factors obtained were combined with other demographic, radiological, and spirometric variables to discover a causal graph using the fast causal inference (FCI) algorithm. This method exploits conditional independencies between observational measurements to determine potential causal/effect relationships. Finally, to extract COPD subtypes, we employed the single sample network perturbation assessment (ssNPA) algorithm which works by splitting subjects into reference (healthy) and perturbed (disease) groups, generating a causal graph for that reference group, and predicting outcomes for the perturbed group based on the connections found in the reference causal graph. Poor predictions from the reference group indicate a perturbation in the causal graph caused by the disease. Individuals within the disease group are then clustered by similarities in their perturbations. The reference group was defined according to lung function and was comprised of 97 subjects.
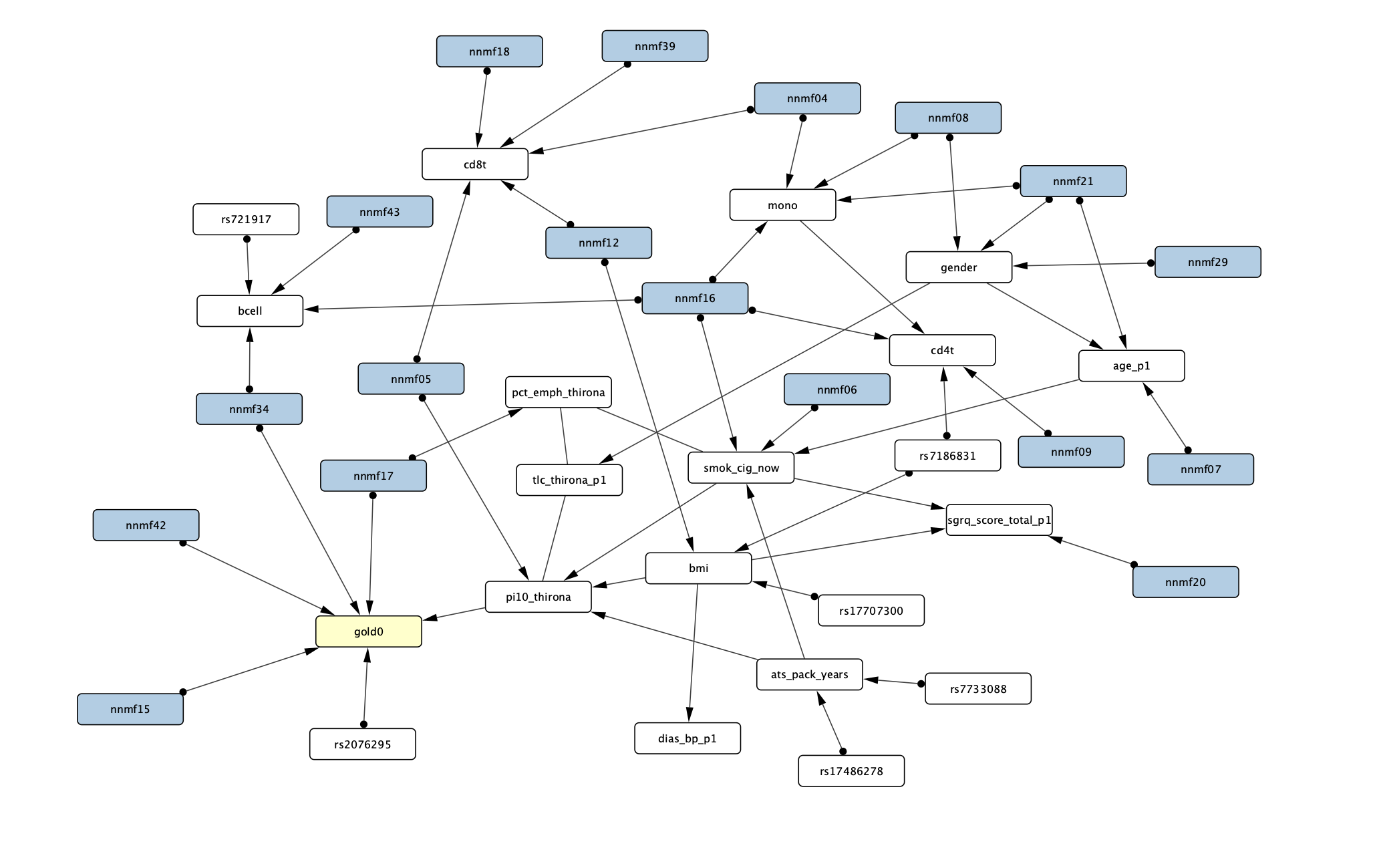
Results. Using a 5-fold cross-validation scheme, a total of 45 NMF factors were determined from the methylation data. Combined with other observational data provided by COPDGene, the FCI algorithm identified 4 NMF factors that directly link to early COPD progression (Figure 1). Within the NMF15 variable, gene ontology analysis identified biological mechanisms associated with positive regulation of CD8 positive T cell proliferation as well as natural killer cell mediated cytotoxicity. ssNPA identified 8 clusters across COPDGene subjects. Within the two largest clusters, we found a significant difference in 5 -year survival (p=0.0037). This survival difference is likely explained by DNA methylation as no difference in COPD severity (p=0.41) was found.
Conclusions. This analysis suggests that epigenetic perturbations caused by smoking exposure play a significant role in the progression of COPD later in life. These long-lasting changes to the DNA methylome affect how COPD progresses and causal inference algorithms can identify subtypes based on these perturbations. Similar analyses need to be performed in external cohorts to verify these COPD methylation subtypes.