2025 AIChE Annual Meeting
(407g) Unraveling Temperature-Dependent Free-Energy Landscapes and Surface Dynamics in Methane Activation on Ni(511) Via Machine Learning and Enhanced Sampling
Authors
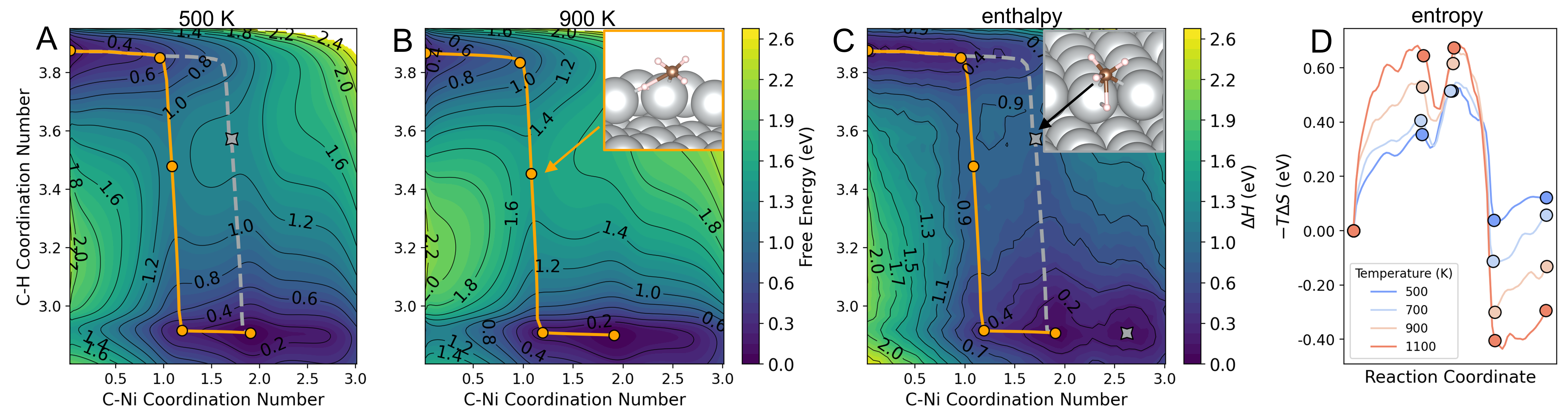
As methane approaches step-edge Ni atoms, its entropy drops, reflecting reduced mobility. Once the system surpasses the transition state leading to C–H cleavage, the mobility of adsorbed species increases, resulting in a notable entropy gain (Fig. 1D). Further, as the methyl (*CH₃) species binds to additional surface nickel atoms, its mobility diminishes again, lowering the entropy. This trend persists as *CH₃ diffuses between atop, bridge, and hollow sites. Similarly, for the [CH₃–H]‡ transition state, an alternative activation route involving two Ni atoms at a step-edge bridge site competes with the atop route. At low temperatures, both routes are identifiable; however, the bridge-site mechanism vanishes at temperatures beyond 700 K, reflecting entropic penalties against binding geometries involving more surface Ni atoms (Fig. 1A-C). Additionally, we highlight that active sites are neither fixed nor strictly localized at step edges, where thermal fluctuations and entropic effects promote activation at terrace sites as well at higher temperatures. By integrating ES with an accurate MLIP, this work provides a rich molecular picture of methane activation under realistic, elevated temperatures on Ni(511). Beyond static DFT insights, we demonstrate the potential for efficient, dynamic modeling of catalytic systems.