2025 AIChE Annual Meeting
(253e) Tunability of Local Structure and Spin States in Governing ORR Activity of M–N–C Catalysts
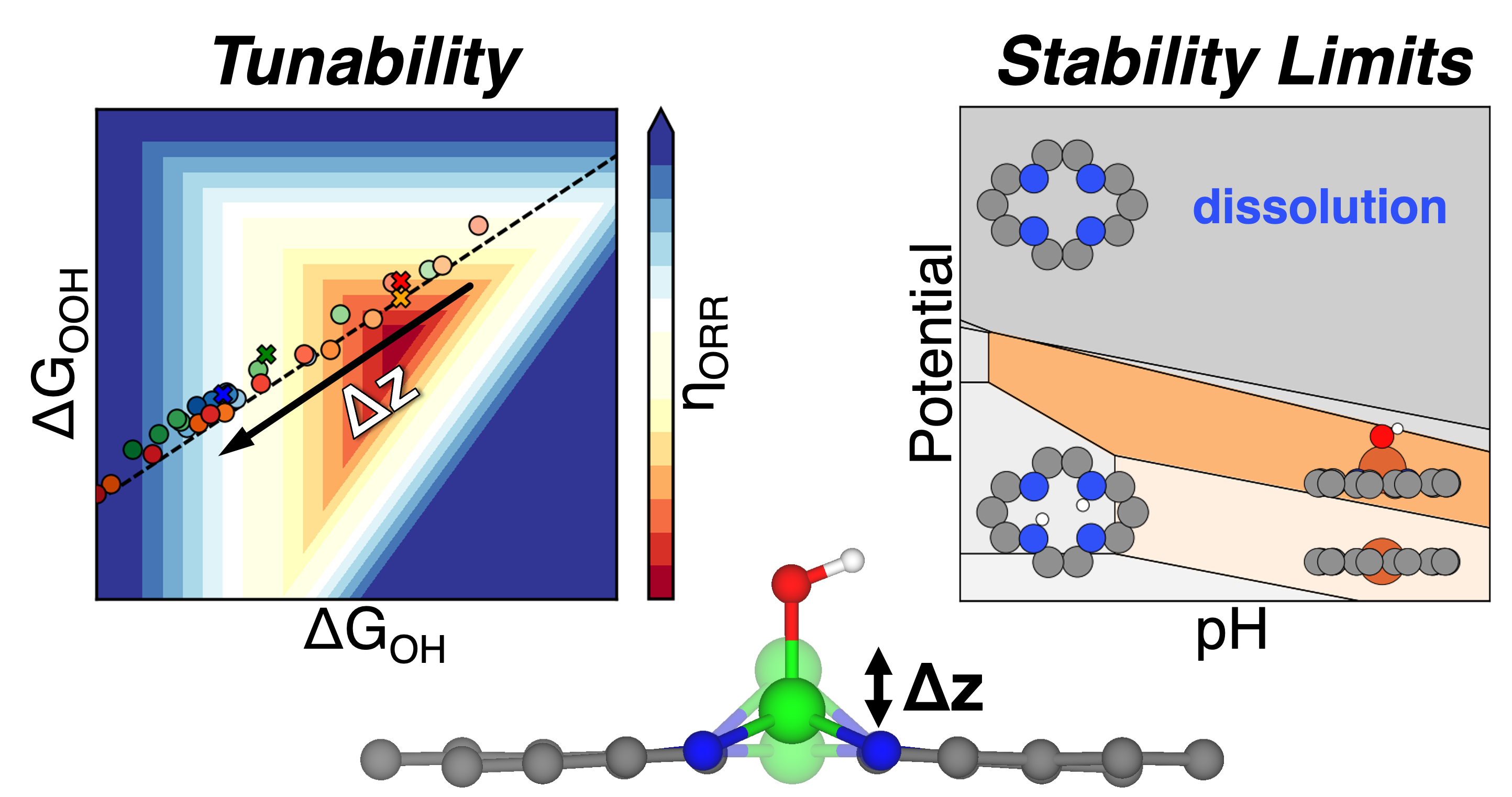
Using density functional theory, we investigated 22 transition metals and identified six (Mn, Fe, Co, Ni, Mo, W) that exhibit displacement-dependent spin transitions, forming double convex hulls in their formation energy profiles. These spin state changes result in non-linear trends in adsorption energies—particularly for *O and *OH—because the relationship between displacement and binding strength changes with spin configuration. This leads to notable deviations from conventional linear scaling.
For the oxygen reduction reaction (ORR), adsorption energies generally strengthen with increasing displacement. As a result, the optimal displacement direction (increase vs. decrease) depends on the catalyst’s position on the volcano plot: left-branch systems like Mn–N–C and Fe–N–C benefit from decreased displacement, while right-branch systems like Co–N–C and Ni–N–C are enhanced by increased displacement. Co–N–C achieves a minimum ORR overpotential of 0.25 V. For the oxygen evolution reaction (OER), displacement similarly modulates adsorption energetics, but yields non-monotonic behavior due to discontinuous changes in spin states. However, stability constraints—such as metal dissolution under oxidative or acidic conditions—limit the tunability window for OER.
This work establishes a mechanistic link between local geometry, d-orbital splitting, and catalytic activity in M–N–C catalysts. By elucidating how displacement-induced spin transitions govern adsorption energetics, we provide a framework for designing spin- and geometry-tunable active sites with optimized performance.