2025 AIChE Annual Meeting
(252c) Structural Evolution of Au-Pd Alloys in the Presence of CO
Authors
Conor Waldt - Presenter, Purdue University
Sucharita Vijayaraghavan, University of Illinois, Urbana-Champaign
Hung-ling Yu, Virginia Tech
David W. Flaherty, University of Illinois At Urbana-Champaign
Ayman M. Karim, Virginia Polytechnic Institute and State University
David Hibbitts, University of Florida
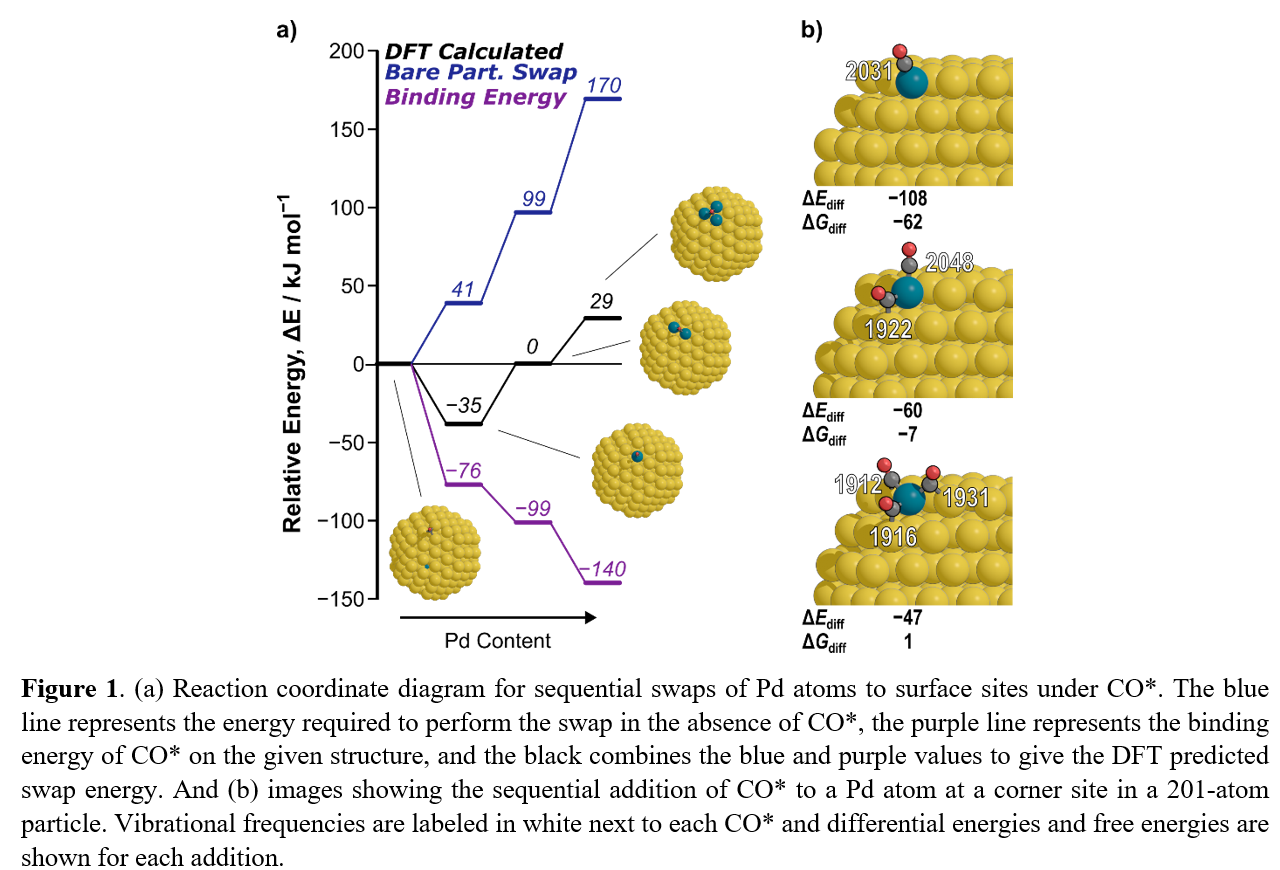
Under vacuum, Pd atoms prefer to be in the subsurface of a nanoparticle and isolated from each other in Au.1–2 However, AuPd catalysts can be restructured in the presence of adsorbates, in particular CO*. Density functional theory (DFT) results on slab models show that CO* can bring Pd atoms from the subsurface to the surface or bring together two isolated surface Pd atoms to form a Pd-Pd dimer.2 These structural changes are brought about by the strong preference CO* has for Pd over Au. Here, we focus on smaller nanoparticles where the effects of undercoordinated metal atoms must also be considered. We performed an initial test with 3 Pd starting in the subsurface of a 201-atom particle, as is expected with a bare particle. After adding 1 CO*, we move 1 Pd to the surface and see it is an exothermic process (as was observed in literature on slab models)2 (Fig. 1a). However, bringing a second and third Pd to the surface is not favorable with only one CO*. This is not to say that multiple CO* will inherently form Pd ensembles either, as we can also sequentially add CO* to a single Pd at a corner site (Fig. 1b). Also, IR spectroscopy data for a ~1.8 nm AuPd nanoparticle shows growth with Pd content in a broad peak ranging from ~1800–2000 cm−1. This peak is attributed to bridge bound CO*, however both CO* bridged on a Pd-Pd dimer (1880–1895 cm−1) and CO* on a mixed bridge with one Pd shared with other CO* (1912–1922 cm−1) fall within the range for this broad peak suggesting IR alone is inadequate in predicting the structure of these alloy particles (Fig. 1b). Therefore, a more rigorous understanding of AuPd alloying in the presence of CO* is required.
References: 1. Nat. Commun.2021,12,1549 2. JPCC.2025.129,11,5702–5717