2025 AIChE Annual Meeting
(58b) Steering Retrosynthetic Planning Towards Chemical Engineering - a Multi-Objective Approach
Authors
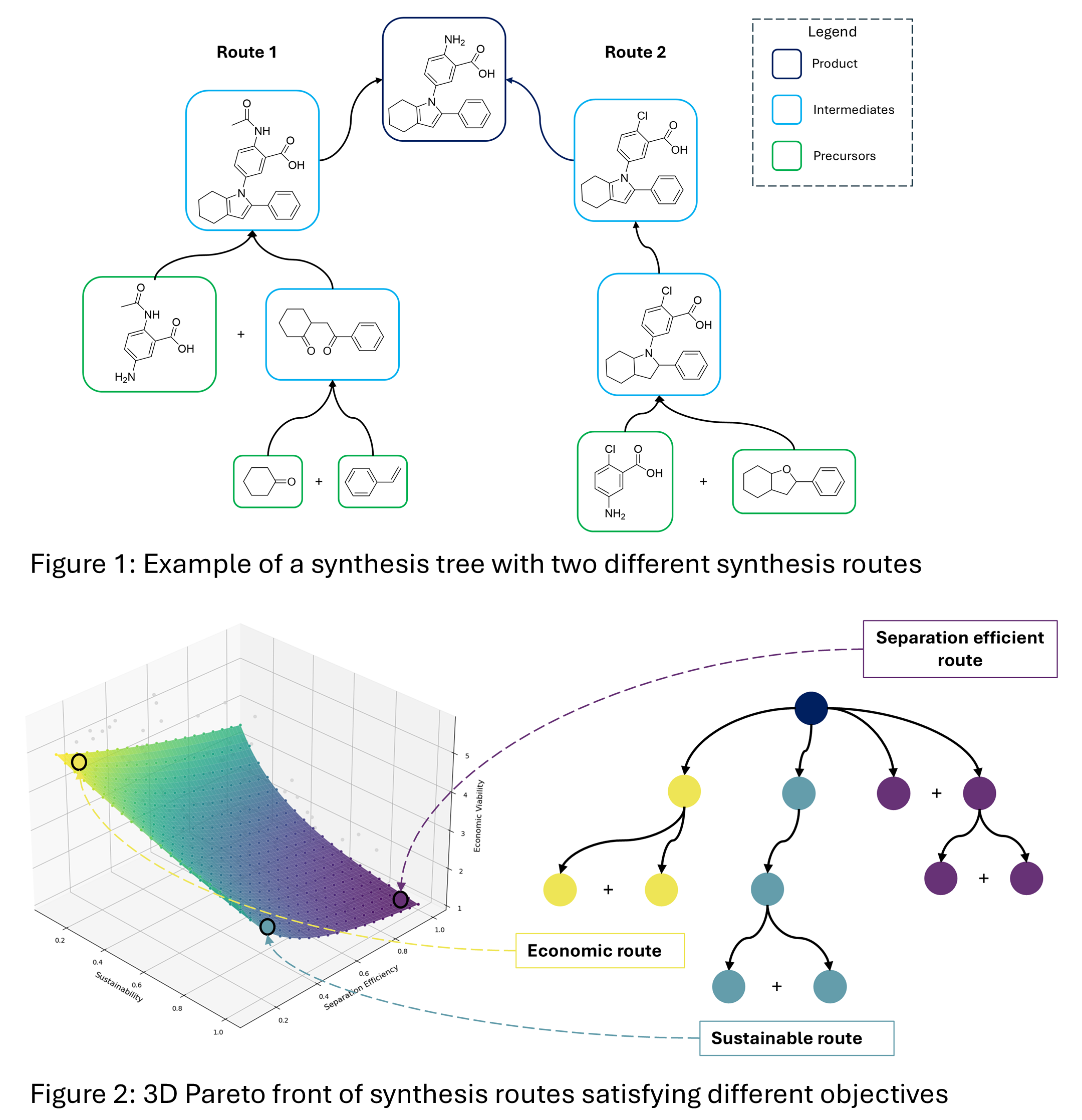
Existing CASP algorithms are usually based on two interacting tools for retrosynthetic planning: i) The single-step chemistry-aware model [2] and ii) The multi-step planning algorithm [3,4]. While the former predicts possible reactions given a molecule, the latter constructs the synthesis route. In other words, the single-step model decides the next step in the chemical search space, while the multi-step algorithm navigates the synthesis path taken in the chemical space. Existing multi-step algorithms are either based on Monte-Carlo Tree Search (MCTS) [3] or A* search [4]. While they differ in methodological details, they both have the same objective: reaching a final goal state at the end of the planning routine. For retrosynthesis, the goal (objective) is to find a set of purchasable building blocks, that when reacted together in several (often consecutive) reactions, will form the desired product. At the end of retrosynthesis planning, the end user is presented with N synthesis routes (in form of a synthesis tree - see Figure 1), which they can choose from.
Existing CASP tools are based on the single objective of finding purchasable building blocks [5]. While this is indeed important, it can be considered more of a search constraint in order to arrive at valid synthesis route; a synthesis route is only considered valid if all reactants in the route are purchasable. By focusing only on the single-objective, the resulting synthesis routes produced by the CASP tool do not consider other important considerations in the realm of chemical engineering such as separation efficiency, sustainability, energy requirements and the overall economic value. While the end-user can select a synthesis route a posteriori from the synthesis tree, the tree itself may not necessarily consist of an optimal route for desired objective.
In this work, we enhance existing CASP algorithms to better serve chemical engineering needs. Our approach combines A* search with multi-objective optimization to discover synthesis routes that simultaneously balance sustainability, separation efficiency, and economic viability. The search process is guided by neural network "value functions" trained offline, which evaluate the promise of partially explored synthesis routes during inference time. This makes it possible to efficiently explore the large chemical search space for multiple objectives while exploiting promising routes. Rather than producing a single optimal solution, our algorithm generates a Pareto front of optimal routes (Figure 2), allowing users to select the synthesis pathway that best matches their priorities and constraints.
Our results demonstrate the superiority of our multi-objective approach over single-objective A* algorithms. The Pareto front returns diverse synthesis routes optimized for specific goals: routes with minimal global warming potential, pathways designed for efficient separation (e.g., featuring reactants with distinctly different boiling points), or economically optimized routes using inexpensive starting materials. Importantly, our method naturally produces greater chemical diversity across routes: a highly valued attribute among chemists. By embedding multiple objectives directly into the search algorithm rather than applying selection criteria afterward, we ensure that all generated routes satisfy the desired engineering constraints. This approach serves both academic researchers and industrial clients with varying priorities, eliminating the need for suboptimal post-hoc filtering and allowing for direct integration with autonomous systems.
References:
[1] Connor W. Coley, William H. Green, and Klavs F. Jensen. Machine Learning in Computer-Aided
Synthesis Planning. Accounts of Chemical Research, 51(5):1281–1289, 2018. ISSN 0001-4842.
doi: 10.1021/acs.accounts.8b00087.
[2] Shuan Chen and Yousung Jung. Deep retrosynthetic reaction prediction using local reactivity
and global attention. JACS Au, 1(10):1612–1620, 2021. ISSN 2691-3704. doi: 10.1021/jac-
sau.1c00246.
[3] Marwin H. S. Segler and Mark P. Waller. Planning chemical syntheses with deep neural networks
and symbolic AI. Nature, 555(7698):604–610, 2018. ISSN 1476-4687. doi: 10.1038/nature25978.
[4] Binghong Chen, Chengtao Li, Hanjun Dai, and Le Song. Retro*: Learning retrosynthetic
planning with neural guided a* search. In The 37th International Conference on Machine
Learning (ICML 2020), 2020.
[5] Kevin Yu, Jihye Roh, Ziang Li, Wenhao Gao, Runzhong Wang, and Connor W. Coley.
Double-ended synthesis planning with goal-constrained bidirectional search, 2024. URL
https://arxiv.org/abs/2407.06334.