2025 AIChE Annual Meeting
(86e) Sequential, Multiplexed Immunofluorescence Imaging of Live Cells Based on DNA-Mediated Reversible Attachment/Detachment of Fluorophores with Antibodies
Proteins play essential roles in the physiological environment, and their irregularities are often associated with diseases. In our body, numerous proteins exhibit specific distribution patterns on/within cells, through which they function in a coordinated manner. Therefore, a comprehensive understanding of the spatial distribution of numerous proteins is crucial for elucidating the mechanisms underlying biological phenomena and diseases. Immunofluorescence imaging has been widely used to observe protein expression patterns owing to its high sensitivity and specificity. However, the current issue is that existing techniques can only image a few proteins simultaneously, typically fewer than five, due to the spectral overlap of fluorescent dyes, which hinders the comprehensive analysis of various proteins. Therefore, improvements in traditional imaging techniques are required.
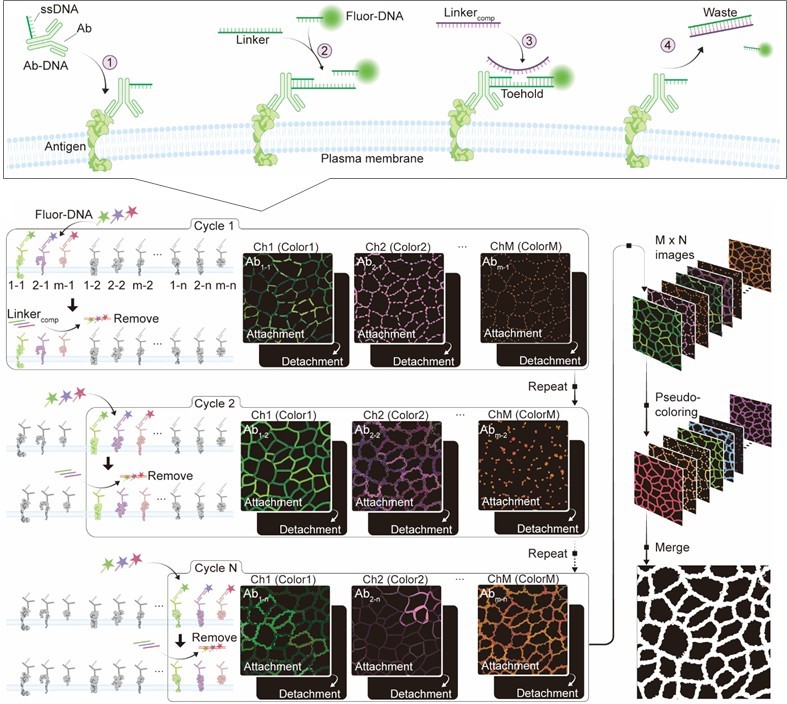
Sequential imaging methods have been considered to have great potential for overcoming the spectral overlap of fluorophores. In each cycle, M target types were simultaneously stained using fluorophores, and after imaging, the fluorescent signals were quenched by a “destaining process” to complete one staining cycle. By repeating this cycle N times, M × N types of protein markers can be imaged from a single sample. The key challenge of this sequential imaging technique is the destaining process. Destaining generally involves harsh chemical treatments, such as reducing agents or oxidation chemistry, which can alter cell structure, induce cytotoxicity. Presumably, owing to these limitations, the existing sequential imaging techniques have only been applied to fixed cells.
Unlike methodologies that use harsh chemical reagents as described above, the biocompatible nature of the hybridization reaction makes DNA an ideal tool for mediating dynamic processes in live cells under physiological conditions. For example, toehold-mediated strand displacement (TMSD) allows for the rapid exchange of nucleic acids within the DNA complex through reconstruction, leading to facile dissociation of the existing structure. In this study, we developed a sequential protein imaging method for living cells by employing DNA as a detachable linker to bind antibody (Ab) with fluorophores [1]. A single-stranded DNA (ssDNA)-modified Ab (Ab-DNA) was bound to the target protein marker in the cells. Fluorophore-conjugated ssDNA (fluor-DNA) was then added to label the Ab-DNA via DNA double helix formation. After imaging, the fluorophore was detached from the Ab via TMSD by adding perfectly complementary ssDNA to the linker (Linkercomp). In this single staining cycle, M non-interfering fluorophores can be used to stain the M markers simultaneously. By repeating this cycle N times, M × N protein markers were imaged in the merged image.
Experimental
DNA modification of antibodies was carried out based on a previously reported method [2], using succinimidyl-[(N-maleimidopropionamido)-diethyleneglycol] ester (SM(PEG)2) as a crosslinker. The average number of DNA molecules per antibody is calculated by measuring the concentrations of antibodies and DNA using the BCA assay and OliGreen assay, respectively.
For demonstrating sequential imaging, six DNA modified antibodies (anti-EGFR, CD44, ICAM-1, Integrin β1, E-cadherin, and CD24) were incubated with A431 cells at room temperature for 1 hour. After that, linker and fluorophore-modified DNA corresponding to EGFR and CD44, and ICAM-1 were added to the cells. Imaging was performed using a confocal laser microscope, followed by the addition of corresponding linker-comp for quenching. After that, similarly to the first cycle, linker sequences corresponding to Integrin β1, E-cadherin, and CD24 were added, followed by the addition of fluor-DNA and subsequent confocal laser microscopic observation.
Results and Discussion
DNA modification of antibodies was carried out based on our previous study [2]. The successful DNA conjugation to the antibody was suggested by the shift of absorbance peak from 260 nm to 280 nm. The average number of DNAs conjugated to each antibody was estimated to be 2.9 by the quantifying antibodies and DNA via BCA and Oligreen assay.
To validate the feasibility of DNA-based sequential imaging, we first conducted a single-color, multi-round imaging experiment on live A431 cells. FITC-modified ssDNA was used to sequentially label six different proteins (anti-EGFR, CD44, ICAM-1, Integrin β1, E-cadherin, and CD24) in six distinct staining cycles. Each cycle involved the attachment of FITC-DNA to Ab-DNA, followed by quenching using linkercomp to detach the fluorophore. Confocal microscopy confirmed successful imaging without significant residual fluorescence between cycles, demonstrating the feasibility of multi-round staining for different protein targets.
Then we demonstrated the potential for M × N multi-color staining in both A431 and A549 cells. Three antibodies (anti-EGFR, CD44, and ICAM-1) were labeled simultaneously using three distinct fluorophores (FITC, TAMRA, and Cy5) in the first cycle, followed by a detachment process to quench signals using linkercomp. Afterward, another set of three antibodies (Integrin β1, E-cadherin, and CD24) was labeled with the same fluorophores (FITC, TAMRA, and Cy5) in the second cycle. This 3 × 2 staining process enabled the imaging of six protein markers in two cycles. Confocal microscopy confirmed successful imaging without significant crosstalk or residual fluorescence between cycles. We further quantitatively analyzed the expression levels between the two cell lines and compared them with publications, validating that this method effectively analyzes the differences in multiple protein expression levels in live cells.
Conclusions
We developed a novel method using DNA as a removable linker to achieve sequential imaging of multiple proteins in live cells. In multiple staining rounds, we verified the possibility of repeating over six cycles and using three colors of fluorophores for each round in living cells. This approach provides a new tool for life science, greatly enhancing our ability to study protein interactions and expression patterns in live cells.
Acknowledgement
This work was supported by JSPS KAKENHI, JST PRESTO, and JST FOREST.
References
- X. Li, Y. Maeda, N. Nakamura, S. Ohta (submitted)
- Y. Maeda, N. Nakamura, S. Ohta: Adv. Funct. Mater., (2024) 2315160.