2025 AIChE Annual Meeting
(30h) Rejuvenating Digital Discoveries of Metal-Organic Frameworks: In-Silico Screening, Fine-Tuning Featurization and Diversity-Driven Machine Learning
However, the effectiveness of in-silico screening critically depends on accurate predictions of material properties through machine learning (ML). We further developed the Pore+ descriptors, which enhances geometric characterization by incorporating domain-specific chemical insights. This refined featurization significantly improves both prediction accuracy and interpretability compared to conventional pore descriptors.
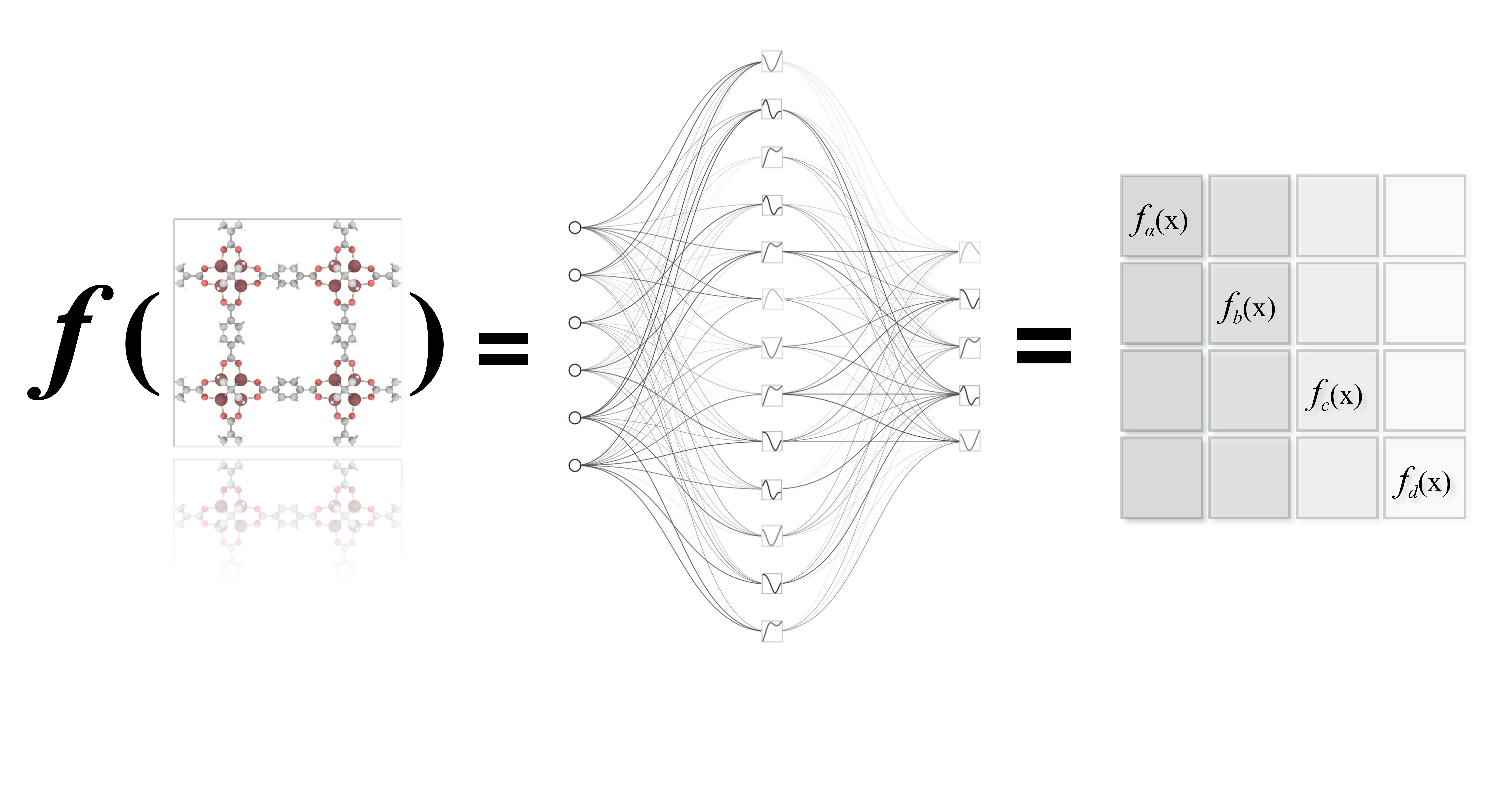
Nevertheless, advanced descriptors alone are insufficient when confronted with diverse datasets or unexplored chemical spaces, as traditional ML approaches often struggle with generalization. To address this challenge, we embraced cross-diversity validation and novel deep learning architecture (e.g. Kolmogorov–Arnold Networks). By integrating insights across heterogeneous datasets, these strategies enhance the robustness and predictive capability of our computational models substantially.
A particularly promising trend is the integration of these digital tools into cohesive, closed-loop pipelines. By unifying RTA-based structure generations, property prediction, and validation steps, we aim to establish an iterative design–evaluate–learn workflows. Conclusively, our works set the stage for a rejuvenated, data-driven digital discovery paradigm for reticular chemistry (Figure. 1), accelerating the virtual exploration and development of next-generation MOFs.