2025 AIChE Annual Meeting
(8f) Reactive Molecular Dynamics Study of the Oxidation Behavior of Copper Nanoparticles
Authors
Anders Hellman, Chalmers University of Technology
Henrik Ström, Chalmers University of Technology
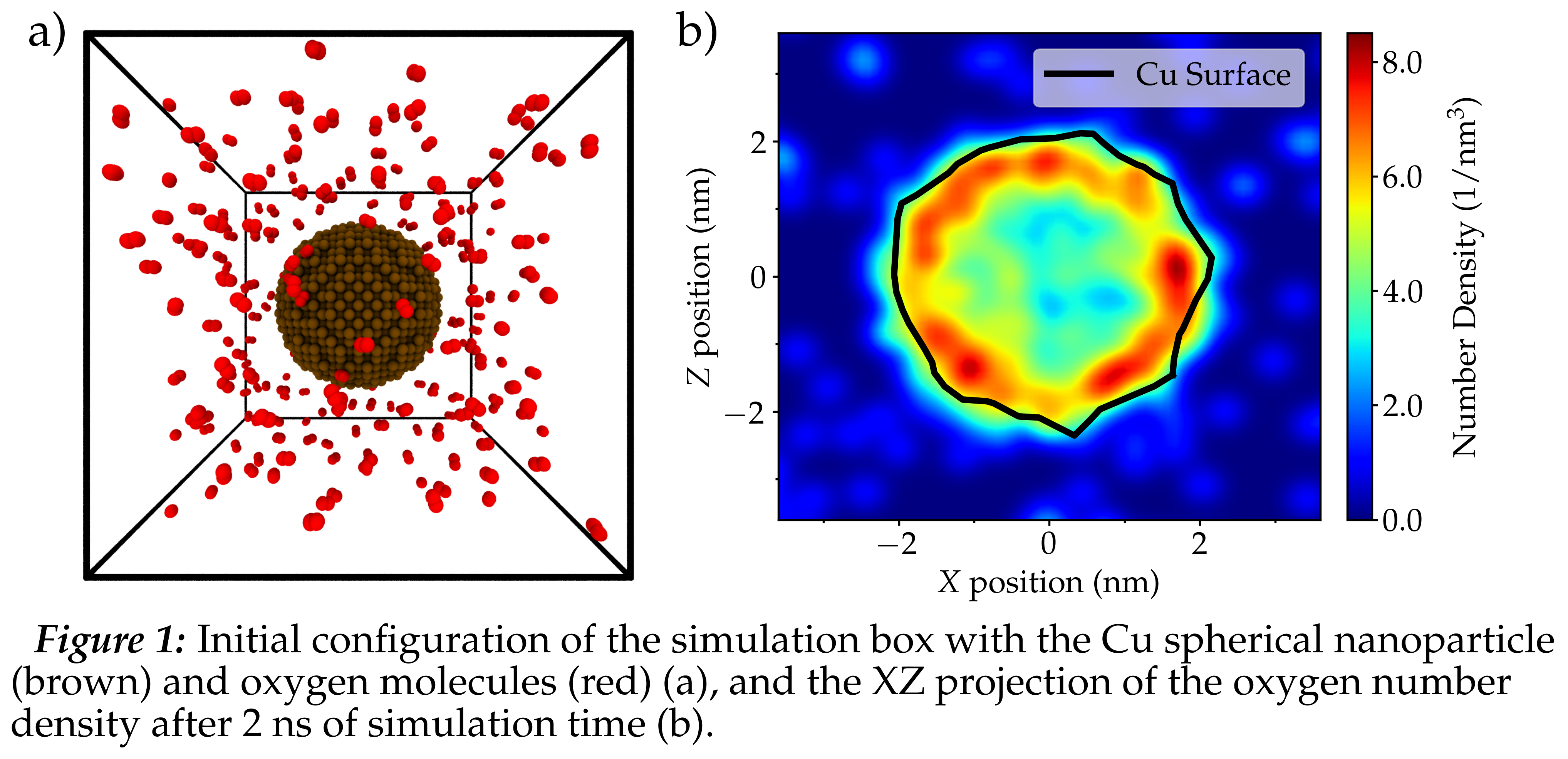
Understanding the oxidation behavior of copper (Cu) nanoparticles is important for applications in catalysis, electronics, and materials science. This study employs reactive molecular dynamics (MD) simulations to investigate the atomic-scale mechanisms governing the oxidation of Cu nanoparticles. By using the reactive force field (ReaxFF)[1], we simulate the interaction of Cu nanoparticles with oxygen under varying conditions, capturing the structural evolution, morphological changes, and oxidation kinetics. We use the Large-scale Atomic/Molecular Massively Parallel Simulator (LAMMPS) to construct and simulate Cu nanoparticles exposed to oxygen environments (Figure 1a). Key parameters, including particle size, temperature, and oxygen pressure, are systematically varied to assess their influence on the oxidation behavior. Structural analysis is performed by evaluating properties such as the radius of gyration, oxidation rate, and density profile evolution (Figure 1b). Additionally, the relative diffusion time of copper and adsorbed oxygen atoms is monitored and analyzed to understand the oxidation dynamics in detail. Our simulations provide key insights into the oxidation mechanisms of Cu nanoparticles, revealing size-dependent and size-independent trends in oxidation behavior. Temperature and oxygen pressure are found to significantly impact oxidation kinetics, leading to distinct morphological changes. Comparative analysis with experimental data from the literature, such as bond dissociation energy, cohesive energy, bond distance, and lattice constant, further validates the computational results and highlights the potential for predictive modeling of oxidation processes in nanoscale systems. This study advances the understanding of Cu nanoparticle oxidation by identifying critical factors influencing structural evolution at the nanoscale. Furthermore, this work underscores the importance of computational approaches in exploring complex chemical and structural transformations in nanomaterials.
References
[1] Senftle, T.P., et al., The ReaxFF reactive force-field: development, applications and future directions. npj Computational Materials, 2016. 2(1): 15011.