2025 AIChE Annual Meeting
(197e) Potential Oscillation Controls Rates and Selectivities of Alkene Epoxidation and Oxygen Evolution Reactions on Au Anodes
Authors
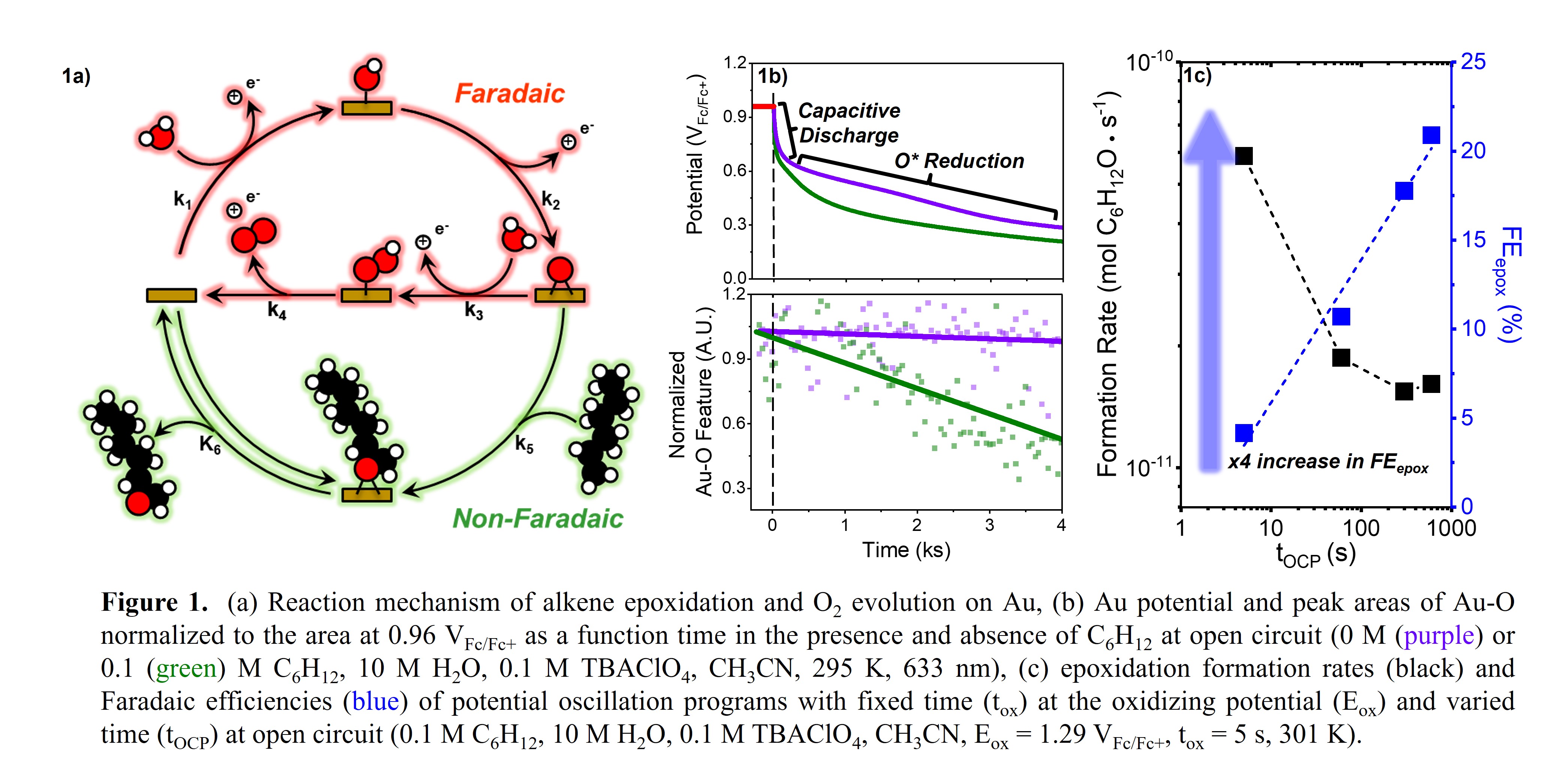
Raman features at 580 cm-1 show that O* species populate the Au surface after polarization to 0.96 VFc/Fc+. Subsequent time dependent OCP measurements display two regimes with distinct kinetic behavior that reflect electrochemical double layer dispersion (0.96-0.85 VFc/Fc+) and O* monolayer reduction (0.85-0.35 VFc/Fc+). Coupled operando Raman spectroscopy supports these interpretations through attenuation of solvated anion and Au-O features (Figure 1b). Rates at which the OCP decays increase with the value of [C6H12] and reflect surface O* consumption. We create potential oscillation programs and find that epoxidation rates and Faradaic efficiencies depend on the potential bounds and duty cycle of the applied square potential waveform. Oscillation programs with periods that equal the time scale of formation and depletion of surface O* maximize epoxidation Faradaic efficiency (Figure 1c). This work demonstrates that process development allows for modulation of selectivity and rates of electrochemical reactions.