2025 AIChE Annual Meeting
(382m) Multiscale Modeling to Guide Catalyst Design, Electrochemistry, and Process Development
Authors
Come find me during my oral talks in AIChE:
Talk-1: Modeling Electroxidation on Molybdenum Carbides: Click here for link to abstract
Talk-2: Developing Interpretable Forcefield for Rare-Earth Elements: Click here for link to abstract
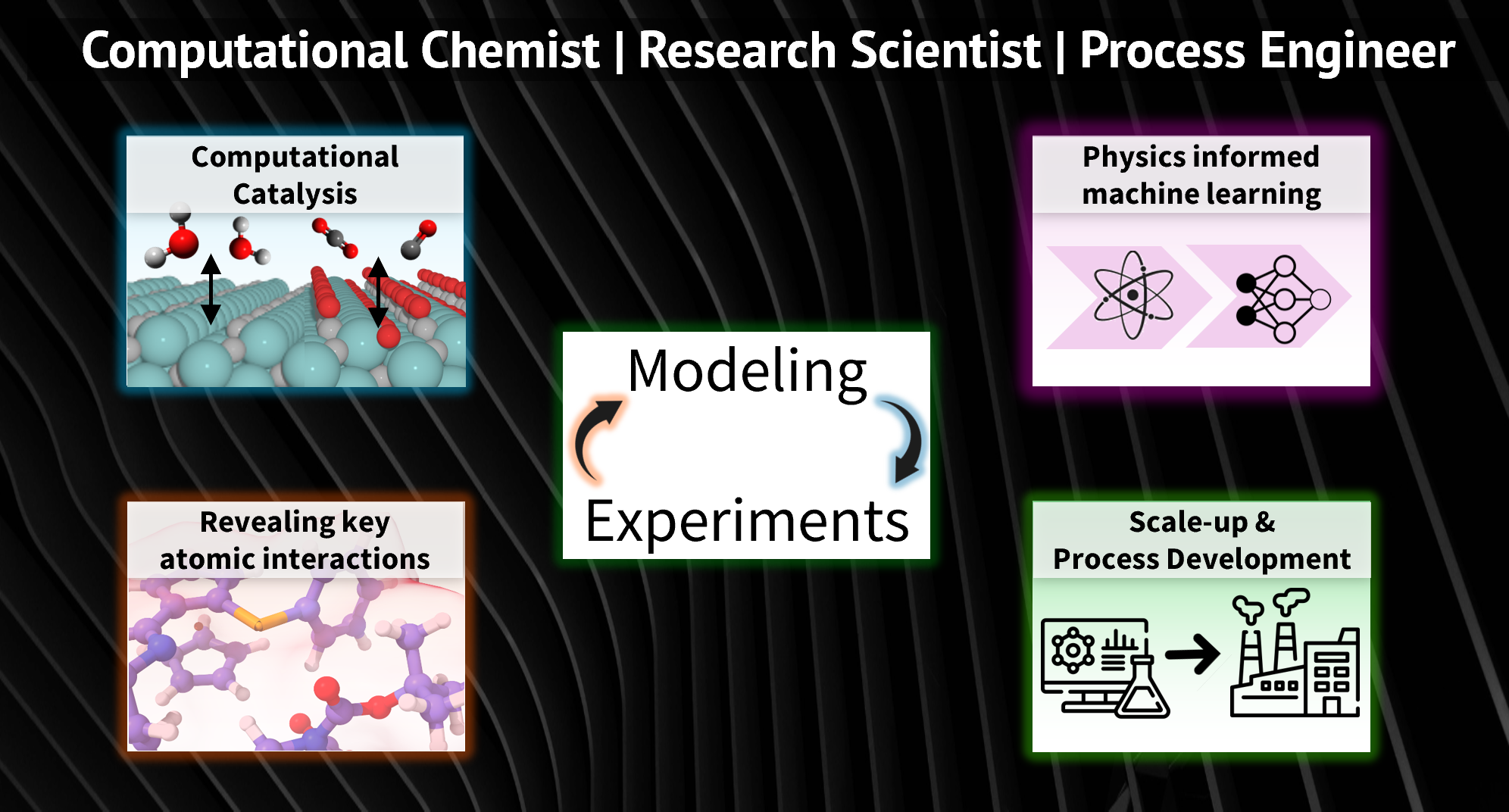
Research Overview:
My research sits at the intersection of modeling and experiments, where I use computational tools to interpret results and guide experimental directions. I have experience in applying a broad range of modeling techniques, from first-principles calculations to process simulations, to derive insights that inform materials and process design. During my PhD, I focused on using first-principles computational modeling to design and understand materials relevant to sustainable technologies.
My work centered on two main areas:
(1) Reducing platinum use by exploring inexpensive molybdenum carbide as an electrocatalyst; See talk-1. In this area, we established a theory-guided surface modeling framework, combining thermodynamics and kinetics, that explains the origins of electrochemical instability for transition metal carbides (TMC). We reveal the important effect of surface oxygen on the stability and catalytic activity of two molybdenum carbide phases, α-MoC and β-Mo2C. This research also reveals how to unlock the platinum-like activity inherent in TMCs, providing strategies for developing more active and selective electrocatalysts [1, 2].
(2) Developing efficient computational methods to drive separation processes for rare-earth and platinum-group metals; See talk-2. In the second area, I developed a computational method that is substantially faster than DFT yet contains key quantum features that describe the solvation of lanthanide cations. This is achieved by using a tight-binding framework with physically motivated functional forms for the individual energy component. Compared to machine learning force fields, the new method requires far fewer parameters, a reduced training dataset, and offers improved interpretability. Additionally, it can aid in designing redox-active materials with enhanced separation performance.
In addition to these projects, I have also contributed to other interdisciplinary projects:
- Identifying atomic-level interactions driving redox-mediated enantioselectivity [3]
- Understanding CH4 overoxidation suppression via surface hydroxyl tuning on WO3 [4]
- Studying surface segregation in multi-metal alloys. Either through application of machine learning or through the synthesis of nanoparticles [5]
Professional Experience:
I also have 1+ years of experience in the pharmaceutical industry as a process engineer within the formulations division. Specifically, I was responsible for 100x scale-up for a drug-coating process as well as improving the tablet spray consistency. In the former, I used mathematical modeling along with CFD-DEM modeling to reveal key operating parameters affecting the quality attributes. In the latter, I developed empirical correlations between the spray atomization parameters to the quality of the spray.
Link to my website : https://gautamankitkumar.github.io/
Linkedin : ankitkgautam
Selected publications:
[1] Gautam, A. K. et al., ACS Catalysis, 2025, doi: 10.1021/acscatal.4c06544
[2] Yu, S., Gautam, A. K. et al., Journal of Materials Chemistry A, 2024, doi: 10.1039/D4TA01746C
[3] Jeon, J., Kappenberg, Y., Gautam, A. K. et al., JACS, 2025, doi: 10.1021/jacs.5c01571
[4] Woo, H., Gautam, A. K. et al., Nano Letters, 2023, doi: 10.1021/acs.nanolett.3c03131
[5] Arif, I., Agrahari, G., Gautam, A. K. et al., Surface Science, 2020, doi: 10.1016/j.susc.2019.121503
Research Summary: