2025 AIChE Annual Meeting
(135b) mRNA-Engineered Dendritic Cell-Based Adoptive Cell Therapy Enhances Cancer Immune Response
Author
Introduction
Immune checkpoint blockade has evolved into a mainstay of current cancer treatment, yet still, only a minority of patients respond. Pre-existing T cell inflamed “hot” tumors portend a successful therapeutic response to immunotherapy, while T cell non-inflamed or immune “cold” tumors respond poorly to immunotherapy. It is believed that immune “cold” tumors suffer from defects in antigen-presentation, a process needed for generating adaptive immune T-cell responses against tumors. Therefore, correcting for defects in antigen presentation would enable the development of anti-tumor T cells and subsequent tumor regression. Dendritic cells (DCs) represent a critical bridge between innate and adaptive immunity and primary antigen-presenting cells controlling T-cell responses. DCs are heterogeneous with discrete cell types and cell states that mutualistically develop with T cells to shepherd T cell effector function in cancer. However, tumors frequently evade the immune system by suppressing DC responses through various means, such as blocking differentiation or maturation, though restoring DCs can re-ignite anti-tumor immunity. However, many current tumor vaccination approaches rely upon the host DC network, which may impair and mitigate anti-tumor immune responses. Thus, DCs have been proposed as a vaccination method to turn “cold” tumors into “hot” T-cell inflamed tumors. Adoptive cell transfer of DCs for cancer vaccination presently has therapeutic implications, with one FDA-approved therapy for prostate cancer and several ongoing clinical trials for diverse cancer indications. While some clinical efficacy has been shown with adoptive DC vaccination, substantial room remains for improvement. Critical concerns for adoptive DC vaccination therapies include i) identification of the target antigen, ii) how the antigen is loaded into DCs, and iii) the activation status of the DC before administration.
One of the latest developments in tumor vaccination is using mRNA-encoded cancer neoantigens to direct adaptive immune responses against tumors. For these treatments, mRNA-encoding neoantigens are encapsulated within lipid nanoparticles (LNP), similar to formulations of COVID-19 vaccines, and often administered intramuscularly, intratumorally, or intravenously. While LNP formulations are generally safe and tolerable for local injections, systemic intravenous administration can be more challenging due to liver toxicity at higher concentrations or repeat injections. Luckily, recent studies have demonstrated that enhancing DC targeting of LNP with in vivo treatment could reduce the dose without compromising therapeutic efficacy. Furthermore, tumors inhibit DC development to evade immunity, suggesting that cancer patients are immune compromised and could be less likely to respond to direct vaccination. Given these and other limitations of current approaches, we set out to explore an alternative strategy.
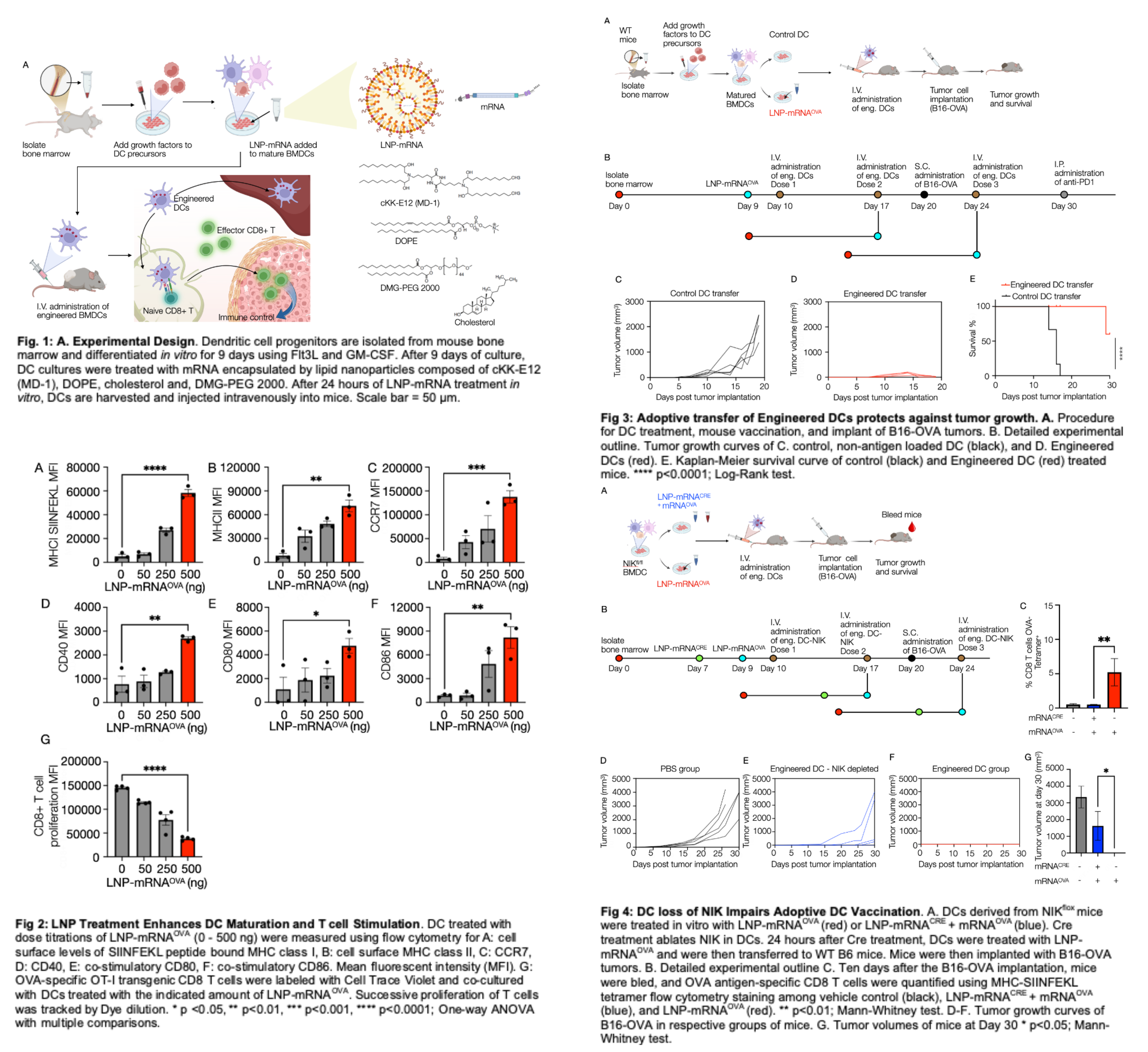
We hypothesized that LNP-mRNA treatment of DC cultures ex vivo before their injection would be much more efficient and safe. We show that ex vivo LNP-mRNA treatment activates harvested DC and strongly enhances antigen presentation and co-stimulation to T cells, using ovalbumin (OVA) as a model tumor antigen (Fig. 1). Transferring these DCs to tumor-bearing mice enhances anti-tumor CD8 T cell immunity without introducing LNP directly to animals and abrogates tumor growth in treated animals. Using this strategy, only scant amounts of mRNA are required to induce robust vaccine effects in mice without systemic organ systems exposure to LNP. Here, we use LNP-mRNAOVA as a model system to show proof of concept for modifying DC. We show that a DC adoptive therapy function can be further modulated by deleting the immune-activating kinase NFkB-inducing kinase (NIK) with LNP-mRNACRE constructs. Overall, we show that LNP-mRNA engineering of DC adoptive therapies is feasible for antigen loading, DC activation, and anti-tumor therapy. These procedures could be employed to enhance DC vaccination therapies in cancer.
Methods
Lipid nanoparticles (LNP) were used as a delivery system for mRNA to bone marrow-derived dendritic cells (BMDC) ex vivo. Different LNPs have demonstrated efficacy in delivering mRNA intracellularly to various immune cells. The LNP preparation used here consisted of cKK-E12 (MD-1), DOPE, cholesterol, and DMG-PEG2000 at a molar composition of 35:16:46.5:2.5. Among the different ionizable lipids possible, cKK-E12 was chosen due to its exceptional biocompatibility, potency, and high mRNA transfection efficacy.
We used modified mRNAOVA and mRNACRE with 5-methyluridine (5moU). This modification has been reported to mitigate interactions with innate immune receptors, thereby slowing clearance and significantly improving efficacy, thus increasing the likelihood of improving the delivery and presentation of mRNAOVA and mRNACRE by dendritic cells. The acidic aqueous phase containing mRNAOVA/mRNACRE was mixed with the organic phase containing the lipids using a microfluidic device. This approach resulted in encapsulation efficiency of 72±8% for mRNAOVA and 68±9% mRNACRE as determined by the Ribogreen assay. The LNP preparations with mRNAOVA had a mean particle size of ~75±6 nm diameter and had a zeta potential of -7.5 mV.
Results
LNP-mRNAOVA leads to antigen presentation ex vivo
To investigate the enhanced activation status of engineered BMDCs with increased concentrations of mRNA, BMDCs were pulsed with LNP-mRNAOVA at three different concentrations (low: 50 ng/500k DCs, medium: 250 ng/500k DCs, high: 500 ng/500k DCs) for 24 hours followed by staining with antibodies. We found a dose-dependent increase in peptide: MHC complex on the cell surface of DCs, showing that antigen can be efficiently expressed and processed following mRNA transfection in vitro on DCs. Antigen presentation was accompanied by dose-dependent enhancement of surface MHC class II, CCR7, CD40, and co-stimulatory ligands CD80 and CD86. To further validate the improved antigen presentation by engineered BMDCs, we co-cultured the LNP-mRNAOVA pulsed BMDCs with OT-I CD8+ T cells. We observed substantial proliferation of OT-I cells, evidenced by dye dilution, with minimal proliferation in control, non-mRNA-dosed conditions. These data show that LNP-mRNAOVA-engineered DC are efficient antigen presenters and can potently stimulate T cell proliferation (Fig. 2).
LNP-mRNAOVA leads to efficient antigen presentation in vivo
Having identified the optimal dosing of LNP-mRNAOVA for ex vivo BMDC engineering, we next cultured bone marrow-derived DC progenitors from wild-type (WT) BL6 mice. It differentiated into DCs by treatment with GM-CSF and Flt3L over 9 days. At day 9, BMDCs were incubated with LNP-mRNAOVA at a concentration of 1 μg mRNAOVA/million cells. Recipient WT BL6 mice received three intravenous injections of the engineered BMDCs (n=4) and non-antigen-educated BMDCs (n=4) as control at seven-day intervals. All mice were then challenged with B16-OVA melanoma tumor cells three days after the second dose of DCs. Twenty days after the first dosing, all the mice received intraperitoneal injections of anti-PD1. Tumor volume increased in the control group at a ~10 times higher rate than the treated group (Fig. 3), and by day 17 post-tumor cell implantation, the tumor volume in the control group was significantly more significant than the treated group, with a statistical significance of p < 0.0001.
DC deletion of NIK impairs vaccination efficacy.
We had previously shown that genetic deficiency of the NFkB-inducing kinase (NIK) impairs immunotherapy response. NIK is most known for its role in controlling non-canonical NFkB activation, and this pathway can be therapeutically modulated to enhance cancer immunotherapy. We hypothesized that we could deliver antigen and genetic modifications to DC to demonstrate the utility of our ex vivo DC mRNA education approach. We thus isolated bone marrow cells from NIKfl/fl mice, differentiated them into DCs, and incubated the DCs with LNP-mRNACRE at a concentration of 1 μg mRNACRE/million cells at day 7 and with LNP-mRNAOVA at a concentration of 1 μg mRNAOVA/million cells at day 9. All the mice were challenged with B16-OVA tumor cells three days after the second dose. Mice receiving NIK competent DCs entirely rejected their tumors, unvaccinated mice exhibited progressive tumor growth, and most NIK deleted DC transfers had substantial tumor growth beyond 1000 mm3, resulting in a significant difference with NIK competent DC group, p < 0.05. Collectively, these data show that mRNA-based engineering of DC can also be combinatorial, as we demonstrate that multiple mRNAs can modify DCs and functionally impact vaccination responses (Fig. 4).
Discussion
LNP-mRNA-based cancer Neo-antigen vaccines are emerging as a new, exciting method to treat cancer. Currently, these approaches rely upon direct LNP-mRNA vaccine administration to the patient, involving systemic dosing. However, it is known that cancer evades immunity by suppressing DC responses and thus undermining T cell immune surveillance. Our approach of ex vivo LNP-mRNA-engineered DCs could enable better vaccination efficacy in immune-compromised or elderly individuals as vaccine responses wane with age. Furthermore, we show that in addition to mRNA-based antigen loading, DC can be functionally modified by mRNA to alter their properties and phenotypic states. This opens up new therapeutic possibilities for engineering adaptive DC vaccines as combinatorial genetic modifications need not be constrained by LNP-mRNA payload capacity and differing in vivo pharmacokinetics of multiple LNP administrations. The limited in vivo half-life of both mRNAs and DCs can reduce toxicity risks, yet they still have long-term effects by priming long-lived effector T cells. Ex vivo mRNA education of DCs and subsequent adoptive transfer does not expose tissues to off-target antigen expression, and transferred DC does not enter the liver. Therefore, the proposed method of vaccination may ultimately result in the most robust vaccination response without side effects.
We further identify that DC-specific deletion of NIK abrogates priming of CD8 T cell responses, consistent with our prior work demonstrating that hematopoietic NIK deficiency blocked the therapeutic response to anti-PD-1 immunotherapy. The main reason for exploring this line of work is to implicate NIK in the DC priming of CD8 T cell responses. This loss of function studies shown herein delineate that NIK is 1) needed for DC to activate CD8 T cell responses and 2) required in DCs for anti-tumor vaccination. Genetic deletion of NIK in DCs in other direct vaccination models likewise impaired CD8 T cell responses yet left CD4 T cell responses intact. This could explain the partially protective phenotype observed with NIK-deficient DC transfer as residual CD4 T cell responses from NIK-deficient DCs could contribute to partial tumor control. We envision that gain of function studies with NIK could potentiate vaccination responses and anti-tumor immunity. While this is a project in itself, the genetic deletion experiments shown here are more straightforward and shed light on specific mechanisms of NIK function on DC immunogenicity. DC subtype may also be necessary for adoptive transfer therapy considerations as transfer of GM-CSF derived DCs, produced predominantly by monocytes, was ineffective in vaccinating murine tumor models.
In contrast, the transfer of type 1 DCs (cDC1), which specialize in antigen cross-presentation to activate CD8 T cells, could confer protective immunity. While our product comprised roughly 35% cDC1, we see strong CD8 T responses elicited by engineered DC transfer. It may be further amplified by selecting cDC1 in our culture conditions and differentiation protocols.
We envision that broadened usage of mRNA-engineered adoptive DC vaccines could open up numerous new therapeutic possibilities for cancer, autoimmune, and infectious diseases. For example, inducing tolerance programs within transferred DCs could enable antigen-specific quenching of autoimmune T cells. Likewise, defining hallmark features of immunogenic DCs could license complete immunity to cancer or infectious disease antigens and could prove useful in deriving new vaccine adjuvants to fine-tune immunity. However, defining the relevant genetic programs to engender tolerance or immunogenicity requires further work. Nonetheless, we establish proof of concept for efficient ex vivo mRNA-engineered DCs capable of triggering potent vaccination and anti-tumor immunity.