2025 AIChE Annual Meeting
(203e) Minimally Empirical, Interpretable Interatomic Potentials for Atomistic Simulations of Rare-Earth Electroseparations
Authors
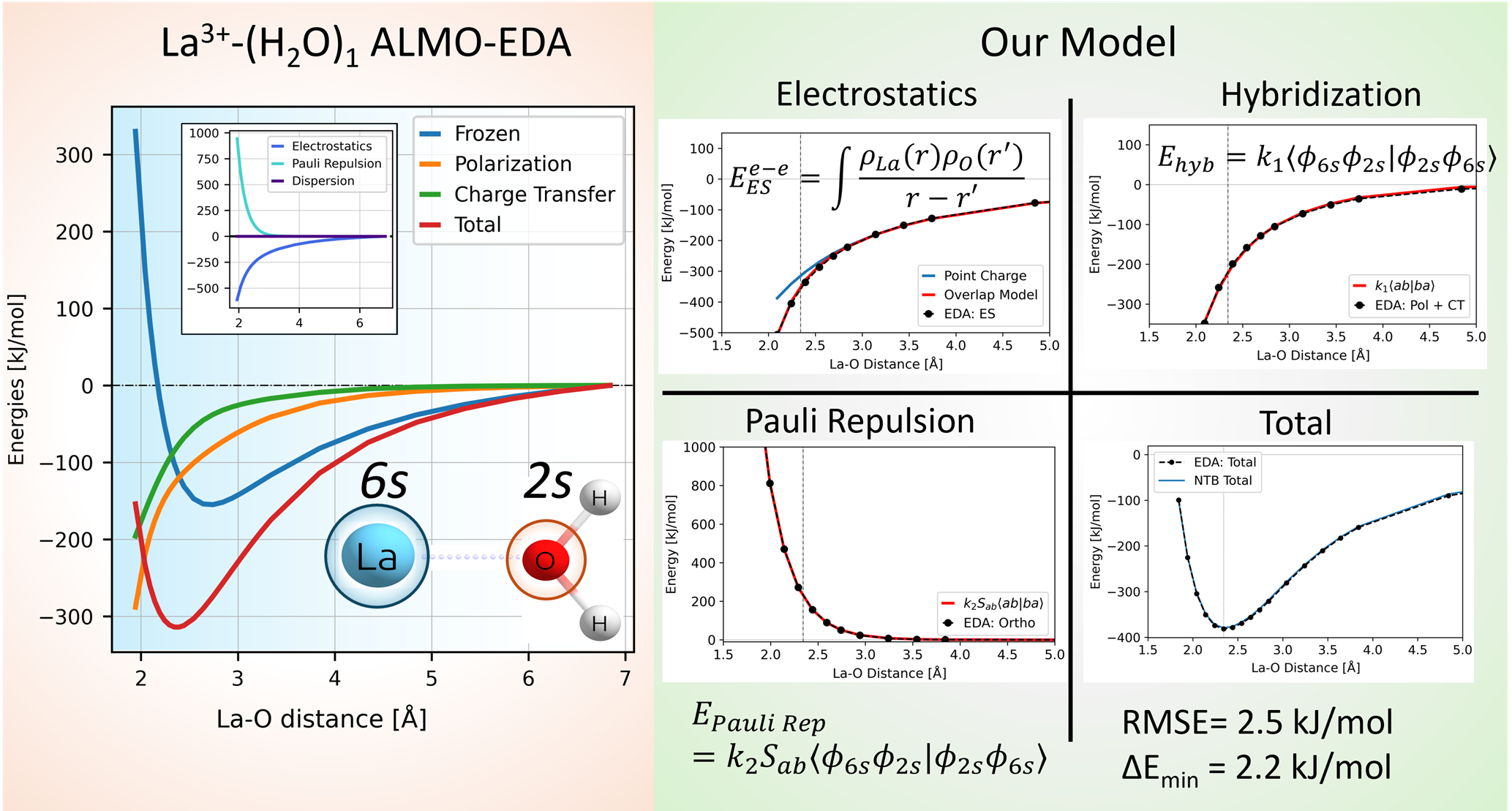
To facilitate long-timescale molecular dynamics calculations, in this work, we are developing a minimally empirical tight-binding theory for aqueous La3+ system. We are fitting physically motivated functional forms to energy contributions obtained from ALMO-EDA (HF-D3BJ/def2-ma-TZVP). For example, to describe permanent electrostatics, we use an atomic orbital overlap dependence attaining high accuracy with minimal changes to orbital exponents. We combine polarization and charge transfer energies to describe hybridization as k<ab|ba>, an expression quantum-mechanical feature. Finally, we model the Pauli repulsion as the product of overlap and the earlier <ab|ba> expression. Our preliminary work on La3+-(H2O) shows excellent performance requiring only a few parameters and only O(10) data points. The simplicity of the functional forms ensures potential transferability to a more complex solvation environments.
By avoiding calculation of expensive 2-electron integrals, our method achieves 500x speedup compared to ALMO-EDA with very little compromise on accuracy. Its fast performance and the limited number of parameters suggests transferability of the model framework for other promising applications such as catalysis.