2025 AIChE Annual Meeting
(526g) Microkinetic Modelling for Electrochemical C-N Coupling By Metal-Organic Materials
Authors
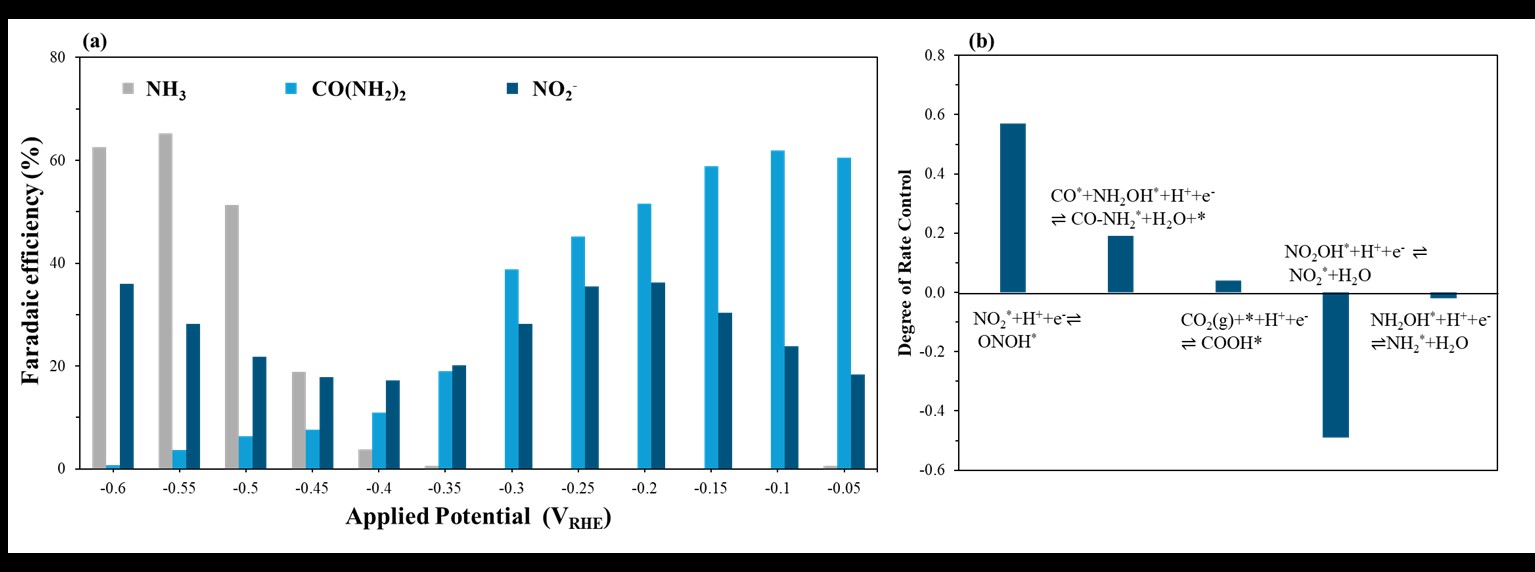
To bridge atomic-scale energetics and macroscopic activity, we integrated reaction thermodynamics and kinetic barriers—calculated via density functional theory (DFT)—into a microkinetic modeling (MKM) framework. The MKM predicts product selectivity and activity as functions of applied potential. Urea and NO₂⁻ formation peak at –0.10 VRHE and –0.20 VRHE, respectively, while NH₃ peaks at –0.55 VRHE, following volcano-type trends. These predictions align well with recent experimental findings [1,2].
Sensitivity analysis reveals that while the initial proton-electron transfers to activate CO₂ and NO₃⁻ are rate-limiting, the *CO–*NH₂OH coupling step dictates selectivity. This step promotes urea formation while suppressing NH₃ and NO₂⁻ byproducts. These insights simplify the complex reaction network into a few key steps, offering a focused pathway for catalyst design.
To generalize this approach, we propose employing active machine learning to identify new candidates across a broader space of metal–BIF (M-BIF) materials. Experimental validation is ongoing to confirm predictions and accelerate the development of high-efficiency electrocatalysts for urea production from waste carbon and nitrogen sources.
Figure 1. (a) MKM predictions for urea, NH₃, and NO₂⁻ FEs. (b) Sensitivity analysis highlighting key rate- and selectivity-determining steps.
References
[1] Yu, X., Xu, Y., Li, L. et al., Nat. Commun. 15, 1711 (2024).
[2] Gerke, C. S., Xu, Y., Yang, Y. et al., J. Am. Chem. Soc. 145, 26144–26151 (2023).