2025 AIChE Annual Meeting
(678h) Machine Learning Accelerated Tailoring of Nanopores for Lithium-Ion Diffusion
Authors
Sauradeep Majumdar - Presenter, École polytechnique fédérale de Lausanne
Swagata Roy, Massachusetts Institute of Technology
Rafael Gomez-Bombarelli, Massachusetts Institute of Technology
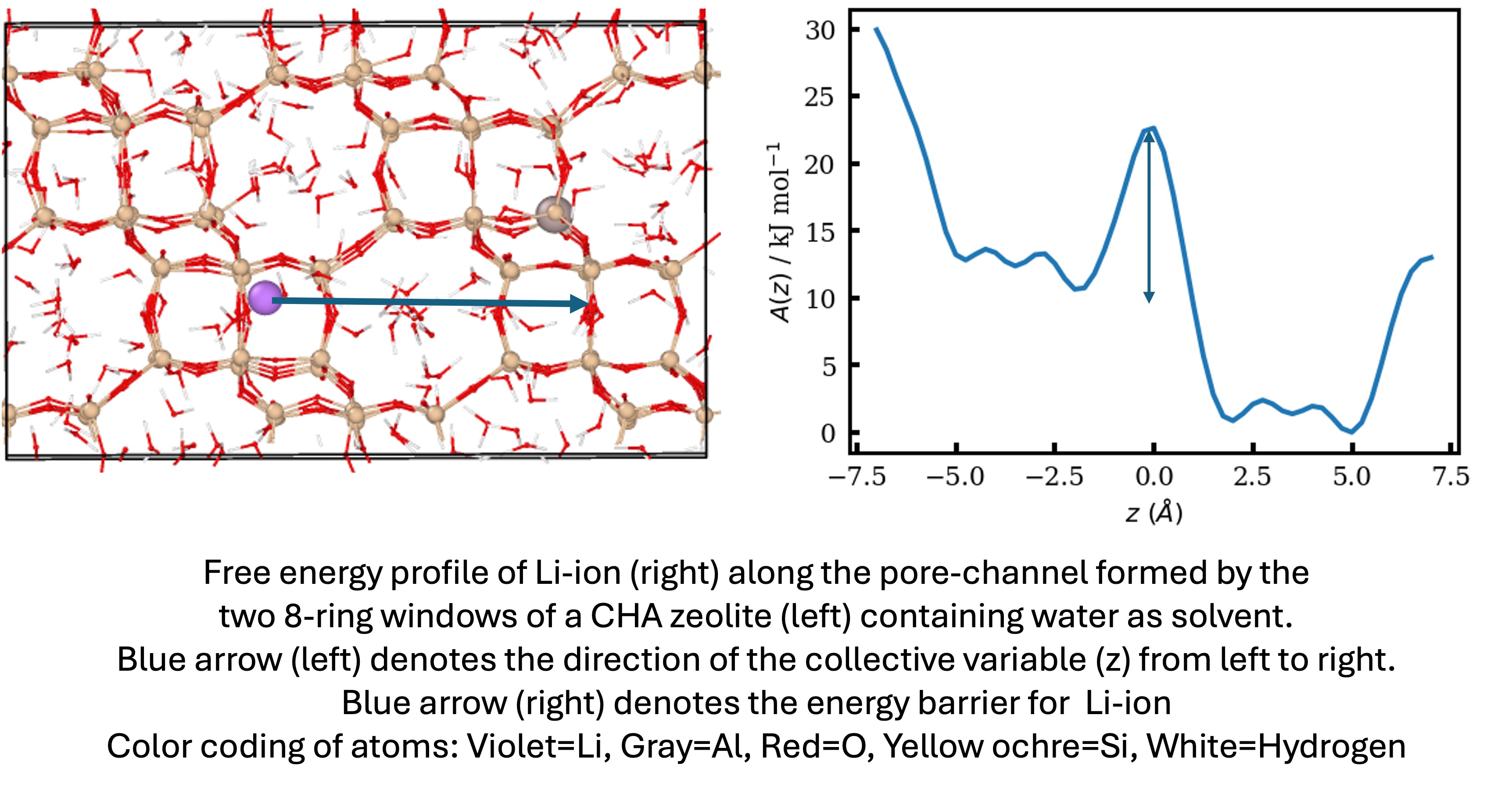
The search for solid-state electrolytes (SSEs) in lithium (Li)-ion batteries as a promising alternative to currently used liquid electrolytes entails the need to understand the ion-transport phenomenon in crystalline nanoporous materials such as zeolites, metal-organic frameworks (MOFs), and carbon nanotubes. In this context, we attempt to understand the energy landscape of Li-ion diffusion in chabazite (CHA) zeolites as a case study, using biased and unbiased molecular dynamics (MD) simulations. To address the challenge of simulating periodic crystalline materials at large length- and time- scales, we used a machine learning interatomic potential, MACE-mp0, to simulate several hundreds of atoms up to several nanoseconds with quantum chemical accuracy. We first justify our choice of the potential and its possible alternatives depending upon the desired accuracy of the property of interest. Free energy profiles of Li-ion obtained from umbrella sampling shed light on the energetic and entropic contributions that lead to the peaks and basins in the energy landscape. Our results underscore the important role played by solvents (H2O in our case) in reducing the energy barrier for Li-ion diffusion by almost 3 times. Subsequent analysis of the Li-H2O solvent coordination environment further shows how the solvent molecules help the Li-ion to cross the energy barriers. Our current work focuses on the systematic tailoring of the CHA zeolite environment, by varying Al distribution, Si/Al ratio, and solvent loading to obtain insights into the effects of nanoconfinement on ion transport through a computational lens. Future work will focus on extending our proof-of-concept to Na-ion transport too, in light of the growing curiosity of the energy industry to move beyond Li-ion batteries. Our work could pave the way and provide insights into the systematic analysis of ion-transport phenomenon into other combinations of SSEs, ions, and solvents, thereby accelerating material discovery for energy applications.