2025 AIChE Annual Meeting
(523d) Interactions between Solvent Molecules and Reactive Intermediates within the Electrochemical Double Layer Dictate Epoxidation and O2 Evolution Kinetics
Authors
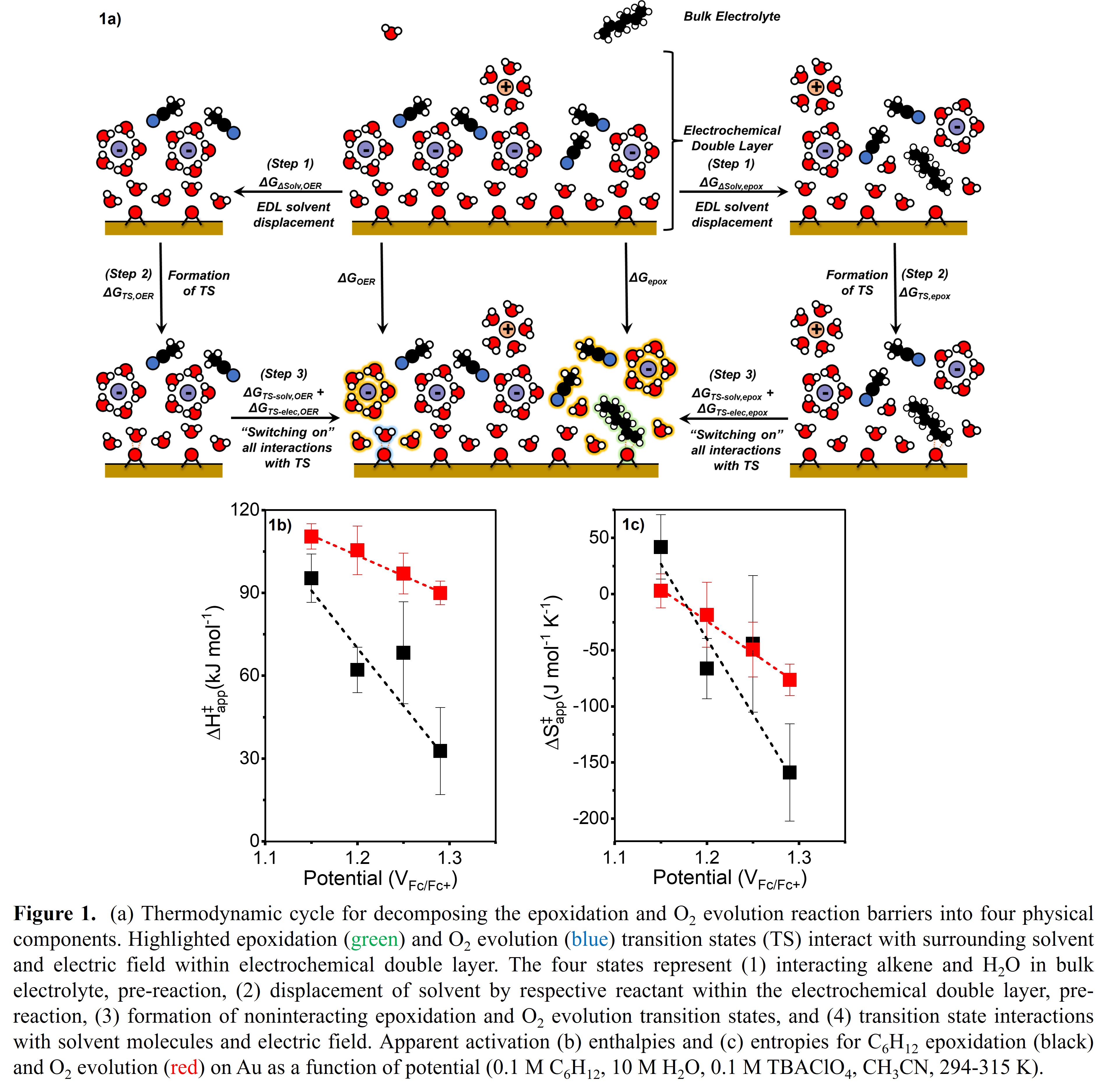
Epoxidation rates and Faradaic efficiencies increase by an order of magnitude and decrease by a factor of two, respectively, as applied potential increases (1.15-1.29 VFc/Fc+). The rate differences do not result from changes in reaction mechanism or mass transfer limitations, but rather changes in transition state stability as quantified, through combination of transition state theory and Butler-Volmer kinetics, by apparent activation enthalpies (ΔH‡) and entropies (ΔS‡). A hypothetical Born-Haber thermochemical cycle deconvolutes the activation barriers to four separate components (Figure 1a). Both the epoxidation and O2 evolution enthalpic barriers decrease with potential (Figure 1b), likely due to increased transition state stabilization from the stronger electric field (step 3, ΔGTS-elec). Both reactions undergo higher entropic penalties at higher potentials (Figure 1c) because the more compact solvent structures within the EDL at higher potentials results in larger entropy losses for the reactants in the bulk electrolyte and requires more energy to displace solvents for the respective transition states (step 1, ΔGΔSolv). We investigate effects of alkene chain length (C3-C12) and electrolyte composition (8-18 M H2O, CH3CN) on apparent reaction barriers to decouple the molecular origins of differences in epoxidation and O2 evolution reaction barriers. The quantification of differences in apparent activation energy barriers induced by EDL solvent structure changes enables informed process and electrolyte engineering for the promotion of epoxidation over O2 evolution.