2025 AIChE Annual Meeting
(641b) Independent Atom Ansatz of Density Functional Theory for Interpretable, Low-Cost Calculations of Potential Energy Surfaces
Author
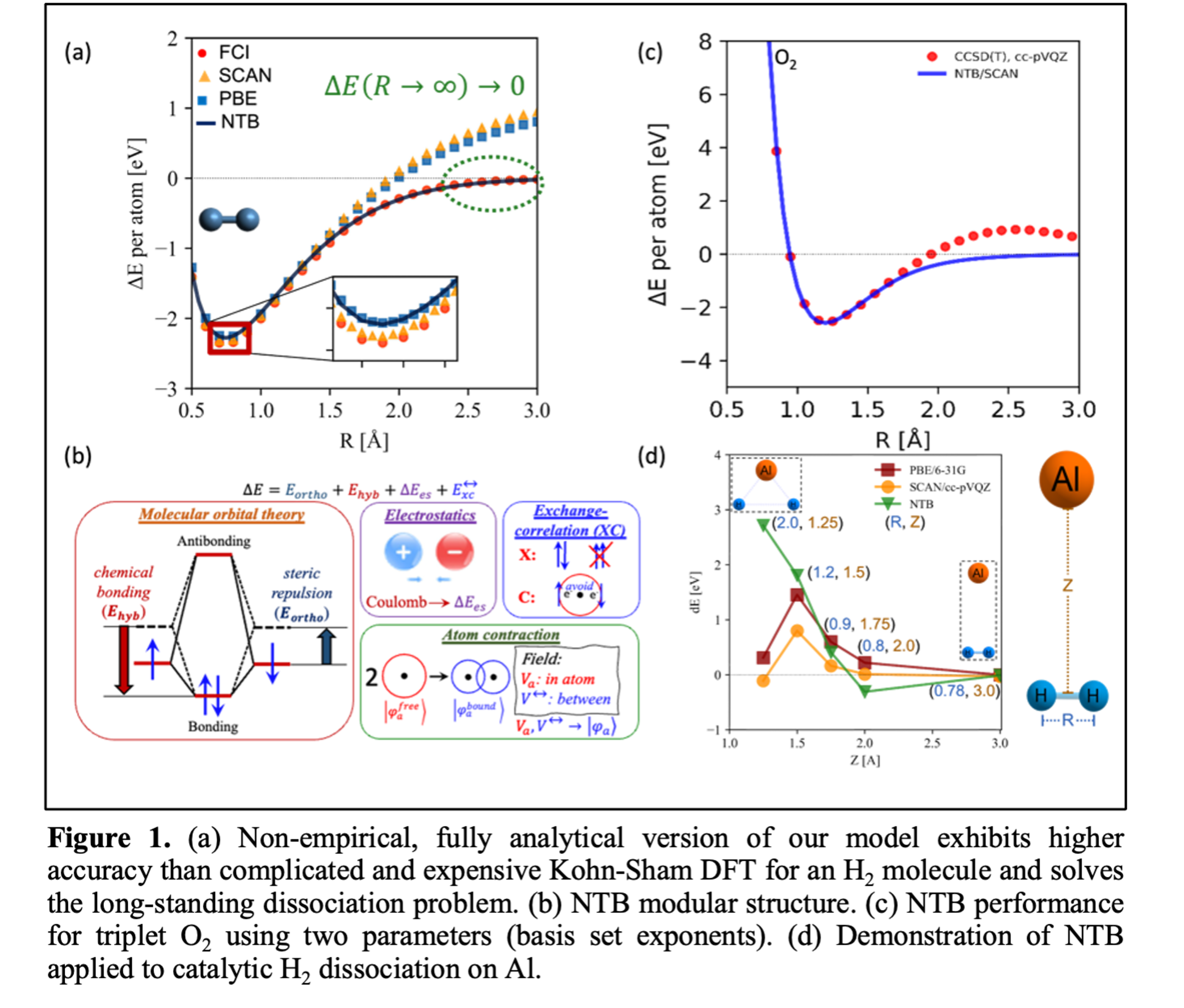
The IA ansatz underpins the nonempirical tight binding theory (NTB), which is parameter-free, describes bond dissociation to free atoms correctly, and incorporates the energy decomposition and charge analyses at no additional cost. NTB predicts the bond energy, bond length, and the vibrational wavenumber of an H2 molecule to be -4.56 eV, 0.743 Å, and 4385 cm-1, respectively, using no parameters, closely matching experimental values of -4.75 eV, 0.741 Å, and 4401 cm-1. The NTB theory has been extended to period-2 diatomics, demonstrating superior accuracy compared to standard Kohn-Sham DFT methods (PBE and SCAN functionals) using at most two parameters: the 2s and 2p exponents of the STO-6G basis set. Ongoing work includes catalytic dissociation of H2 on Al and Pt metals. We anticipate that the independent atom ansatz will enable rapid computational characterization of a wide array of materials and reaction mechanisms, accelerating discoveries in chemistry and catalysis.