
2025 AIChE Annual Meeting
(404e) Hydrogen Bonding Facilitates Transfer Hydrogenation and Hydrogenolysis of Biomass Oxygenates on Transition Metal Catalysts
Catalytic transfer hydrogenation/hydrogenolysis (CTH) using organic hydrogen donors like alcohols or formic acid (FA) offers a milder and safer alternative to molecular hydrogen for upgrading biomass-derived oxygenates. To understand carbonyl hydrogenation mechanisms in sugar-based aldehydes and keto-acids, our previous prototypical model (HCOOH as donor, HCHO as acceptor, Cu(111) catalyst) demonstrated that hydrogen(H)-bonded complexes between intermediates enable a kinetically favorable direct H-transfer, explaining the higher experimental activity observed with FA compared to equivalent H2.
In this study, we theoretically extend these findings to a more realistic system—CTH of 5-hydroxymethylfurfural (HMF) by FA on Pd(111)—using DFT-driven microkinetic modeling. Prior experiments showed that FA as hydrogen source yields higher activity and selectivity toward 2,5-dimethylfuran (DMF). Our results reveal that: (1), hydrogen bonding is critical in both –CHO hydrogenation and –CH2OH hydrogenolysis. The –CHO group undergoes direct H-transfer from FA, while –CH2OH hydrogenolysis proceeds via facilitated C–OH cleavage and H-transfer to the –OH group, forming water. (Figure (a)) (2), H-bonded complexes promote both FA and formate decomposition to CO2 and surface H*, especially in the rate-determining C–H cleavage step. Additionally, the –CH2OH group acts as a hydrogen shuttle to assist hydroxyl H removal from HCOOH. (Figure (b)) (3) in the presence of H-bonding with FA, the DHMF pathway prevails over the 5-MF pathway. (Figure(c)) We posit that, in addition to tuning the catalyst active sites, CTH offers the choice of H-donor as an additional dial to modulate hydrogenation and hydrogenolysis activity. The talk will present a detailed examination of reactions (on the surface and plausibly in the bulk) and the mechanistic consequence of hydrogen bonding in donor-acceptor complexes on transition metals.
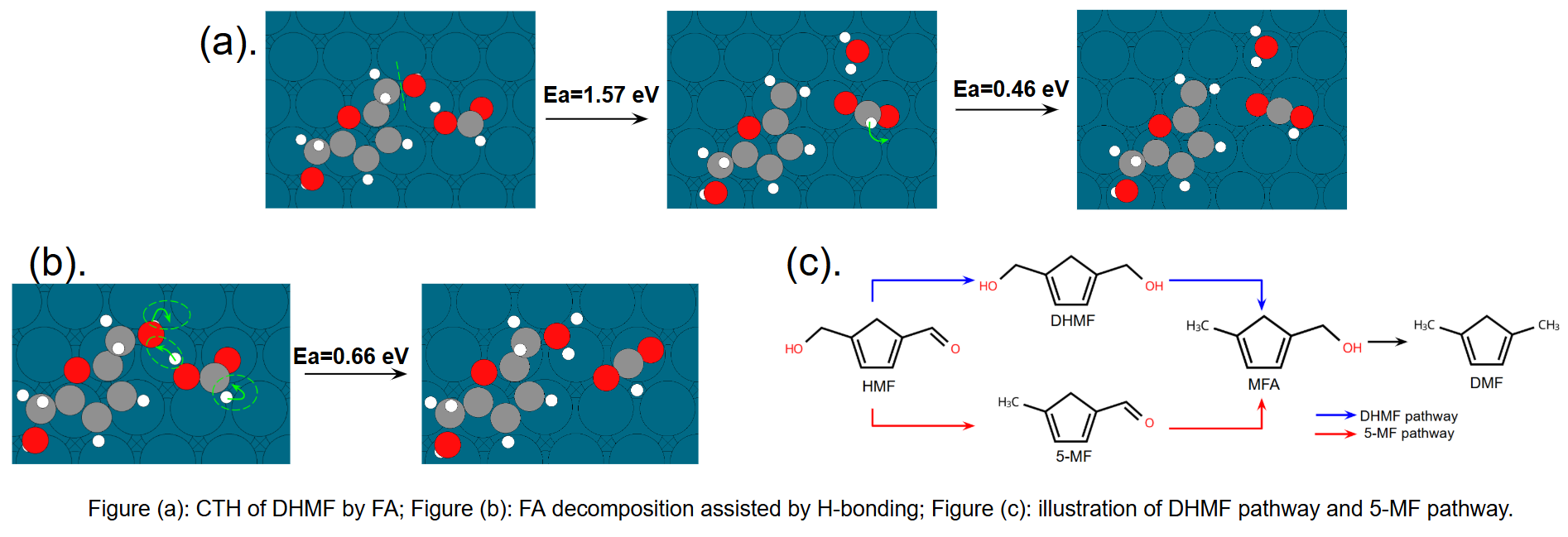
In this study, we theoretically extend these findings to a more realistic system—CTH of 5-hydroxymethylfurfural (HMF) by FA on Pd(111)—using DFT-driven microkinetic modeling. Prior experiments showed that FA as hydrogen source yields higher activity and selectivity toward 2,5-dimethylfuran (DMF). Our results reveal that: (1), hydrogen bonding is critical in both –CHO hydrogenation and –CH2OH hydrogenolysis. The –CHO group undergoes direct H-transfer from FA, while –CH2OH hydrogenolysis proceeds via facilitated C–OH cleavage and H-transfer to the –OH group, forming water. (Figure (a)) (2), H-bonded complexes promote both FA and formate decomposition to CO2 and surface H*, especially in the rate-determining C–H cleavage step. Additionally, the –CH2OH group acts as a hydrogen shuttle to assist hydroxyl H removal from HCOOH. (Figure (b)) (3) in the presence of H-bonding with FA, the DHMF pathway prevails over the 5-MF pathway. (Figure(c)) We posit that, in addition to tuning the catalyst active sites, CTH offers the choice of H-donor as an additional dial to modulate hydrogenation and hydrogenolysis activity. The talk will present a detailed examination of reactions (on the surface and plausibly in the bulk) and the mechanistic consequence of hydrogen bonding in donor-acceptor complexes on transition metals.