2025 AIChE Annual Meeting
(356f) Hybrid Lnp Prime Dendritic Cells for Nucleotide Delivery
Author
Introduction
Most cancers sustain an immunosuppressive microenvironment through elaborated mechanisms that are slowly being unraveled. Among the different immunotherapy approaches, cancer vaccination is of particular interest as it has the potential to elicit systemic and potentially durable anti-tumor effects. Cancer therapeutic vaccines take many forms, such as peptide admixed with adjuvant, adoptive cell therapy of antigen-loaded DCs, or mRNA delivery of encoded neoantigens. Irrespective of the choice of tumor-associated antigens (TAA), immunostimulatory adjuvants are needed to boost vaccine effectiveness. Several different adjuvants have been used, although ample room exists for improvement.
Aluminum compounds are commonly used vaccine adjuvants and are particularly effective at promoting humoral immune responses. Unfortunately, they are less efficacious in inducing cell-mediated immunity, essential for enhanced cancer vaccine efficacy. High-capacity cellular immunity stimulators include modified lipopolysaccharide derivatives (monophosphoryl lipid A) and double-stranded RNA (dsRNA). Synthetic mimics of viral dsRNA include polyinoinosinic–polycytidylic acid (poly I:C), its derivative poly-ICLC (Hiltonol, i.e., poly I:C stabilized with poly-L-lysine and carboxymethyl cellulose), RGC100 and ARNAX among others. These dsRNA mimickers are ligands of the endosomal toll-like receptor 3 (TLR3), most highly expressed in cDC1, cells that are essential mediators of cell-mediated immunity. The mechanism of action of these compounds has been summarized well, and studies comparing their effectiveness are ongoing.
There is considerable interest in potentiating the immune-enhancing effects while minimizing side effects. It should be feasible to enhance dsRNA-mediated TLR3 effects through synergistic and complementary pathway activation in DC. Recent work in tumor-associated myeloid cells has shown that dual NFkB pathway manipulation effectively improves tumor control. This prior work used a 37 nm carbohydrate platform (CANDI) to deliver multiple small molecule immune modulators to tumor-associated macrophages and, to a lesser degree, to DCs. Here, we reasoned that an analogous strategy could be developed for dendritic cell-targeted vaccines but would have to be based on lipid nanoparticles (LNP) to improve dsRNA delivery. LNP has become the nanomaterial of choice for the delivery of nucleic acids, given their considerable payload capacity, favorable pharmacokinetics, and ability to deliver cargo inside target cells. Through recent COVID-19 vaccine efforts, we have a much better understanding of nucleic acid delivery composition.
Here, we describe the synthesis and testing of a new hybrid LNP that contains i) poly I:C and ii) an LNP internal, second nanoparticle as a “sponge” for a minor molecule cIAP antagonist to enhance DC stimulation (Fig. 1). We show the synthesis of the “nanoparticle in nanoparticle” concept and that this approach effectively controls murine colorectal tumors. This novel hybrid LNP can lead to vastly enhanced efficacy over LNP/poly I:C alone.
Methods
We conceptualized a hybrid nanoparticle consisting of a lipid shell, internal poly I:C as a TLR3 immunostimulant, and a smaller, cross-linked cyclodextrin nanoparticle (CANDI) with a therapeutic payload of a cIAP inhibitor. CANDI was synthesized by dissolving succinyl-β-cyclodextrin in MES buffer and activated with N-(3-(dimethylamino)propyl)-N′-ethyl carbodiimide hydrochloride (EDC) and N-hydroxysuccinimide NHS. A solution containing L-lysine in MES buffer (1.5 mL) was added in a drop-wise matter. To render the surface of the nanoparticles hydrophobic (L-CANDI), 3 mg palmitic acid N-hydroxysuccinimide ester was added to 50 mg CANDI. To internalize the CANDI as a reservoir for the model cIAP inhibitor LCL-161, we modified the CANDI surface with palmitic acid to yield L-CANDI. The current research was designed to optimize LNP synthesis, characterize different preparations, and perform in vivo efficacy and mechanistic studies in mouse cancer models.
The LNPs were designed using well-established components and molar ratios resembling those of published LNPs. Initial feasibility studies were conducted to optimize poly I:C and L-CANDI carrying LCL-161 encapsulation. The final preparation consisted of C12-200, DSPC, cholesterol, and DMG-PEG2000 at a molar composition of 35:16:46.5:2.5. We synthesized different LNP formulations to optimize the payloads. LNP14 only contained poly I:C (0.1 mg/mL), LNP15 only contained L-CANDI (25 mg/mL), LNP16 contained poly I:C (0.1 mg/mL) and L-CANDI (25 mg/mL), and LNP17 contained poly I:C and LCL-161 (0.1 mg/mL) loaded L-CANDI. LNP18 is a fluorescent version of LNP17. The different LNP preparations had a mean particle size between 100-125 nm diameter. The zeta potential was negative for all preparations tested (range -4 to -7 mW).
LNPs demonstrated encapsulation efficiency of 75±5% for poly I:C, as determined using the Ribogreen assay and 18.6±2.2% for L-CANDI, as demonstrated using inclusion complexation assay. We determined 10:1 w/w L-CANDI: C12-200 as the optimal loading ratio, as indicated by complete encapsulation. The optimized LNP17 contained 16% lipid, 1% poly I:C, 82% L-CANDI, and 1% LCL-161 by weight. The overall synthetic yield was 70%. The different LNP preparations had a mean particle size between 100-125 nm diameter. The zeta potential was negative for all preparations tested (range -4 to -7 mW).
Results
Screening for Efficacy
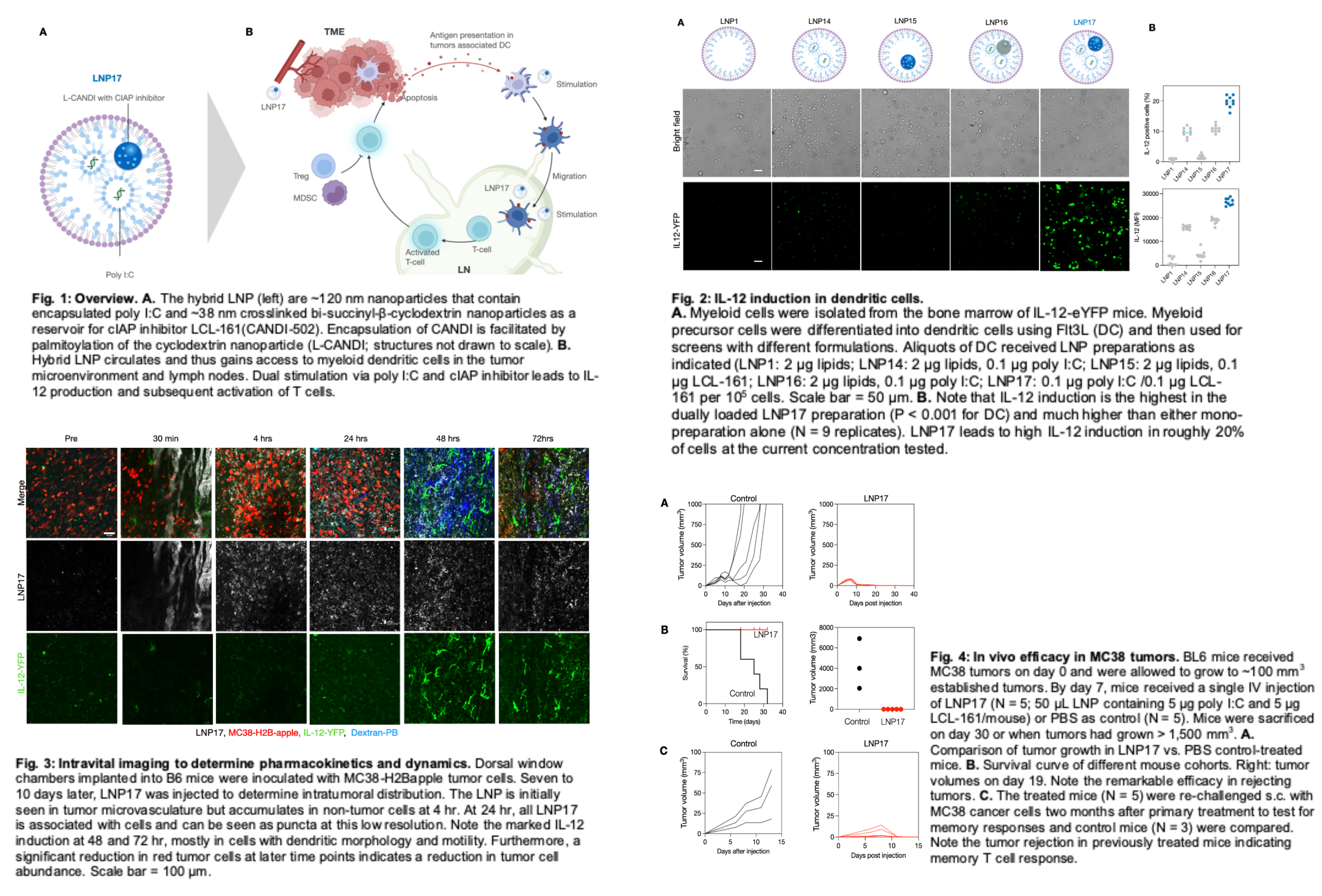
We used IL-12-eYFP donor mice for these experiments and isolated bone marrow cells through femoral flushes. Isolated cells were cultured and differentiated into DCs by treatment with Flt3L over 10 days. Cells were incubated with different amounts of LNP, and IL-12 induction was measured using eYFP fluorescence. We found that LNP17 had the highest IL-12 induction (determined by median fluorescent intensity (MFI) or as % of IL-12 positive cells) (Fig. 2).
Pharmacokinetics (PK) and in vivo efficacy
Having identified the optimum preparation and dose for IL-12 induction in dendritic cells, we next set out to perform in vivo experiments to test the anti-tumor efficacy of LNP17. LNP17 had a vascular half-life of roughly 30 min and accumulated primarily in the liver, spleen, tumor, and lymph nodes 24 hr after administration. To determine the kinetics of cellular accumulation within the tumor microenvironment, we performed intravital microscopy experiments using the MC38 tumor dorsal window chamber model. These experiments allowed single-cell resolution PK/pharmacodynamics (PD) measurement and showed that the materials primarily accumulated in tumor-associated DCs and macrophages (Fig. 3). As expected, IL-12 induction in the cells was time-dependent, with a maximum effect of ~72 hr after IV administration. IL-12 signals were brightest in cells with dendritic morphology, consistent with our prior observations. Interestingly, more extended observation of LNP17-treated tumors showed substantial decreases in tumor cells in imaging experiments that coincided with the induction of IL-12 in the tumor microenvironment.
Having determined the biodistribution and cellular accumulation, we next set out to determine therapeutic efficacy. For these experiments, we implanted MC38 tumors into the flank of BL6 mice. One week after implantation, mice received a single intravenous injection of LNP17 (containing 5 μg poly I:C and 5 μg LCL-161 per mouse). At the injection time, tumor volume in both groups was ~100 mm3. Tumor volumes were subsequently measured by calipers, and survival was recorded. Our data shows complete tumor control in LNP17-treated animals.
Within 1 and 2 days, we observed a ~30% and 80% reduction in tumor volume, respectively, similar to the results we observed by intravital imaging. By day 18, tumors in LNP17-treated mice were eradicated, while control tumors rapidly progressed and reached a volume of ~1000 mm3 (Fig. 4). These results led to a 100% long-term survival of LNP17-treated animals, with all control-treated animals dead 32 days after tumor implantation. To test long-term anti-tumor memory responses, we took complete responders from our earlier LNP17 treatment cohort and re-challenged them subcutaneously on contralateral flanks with MC38 tumor cells 2 months after rejecting their primary tumors. Naive BL6 mice that had never seen MC38 nor LNP17 were used as controls. Naive mice had progressive tumor growth. However, re-challenged mice controlled their tumor growth and had complete eradication of their secondary tumors within two weeks post-challenge, demonstrating a memory response.
We performed a histological analysis of tumors to pinpoint the potential mechanism of action. We examined immune populations in either LNP17-treated or untreated tumors and observed a substantial increase in tumor DCs (CD11c+ cells) and CD8 T cells (CD8+ cells) after LNP17 therapy. Proportions of tumor-associated macrophages (F4/80+ cells) and overall immune cell numbers (CD45+) remained unchanged. We observed co-clustering of DCs and CD8 T cells in LNP17-treated tumors, which was not the case in untreated tumors.
Discussion
The current study shows that high-efficiency hybrid LNP (“nanoparticle-in-nanoparticle”) can be synthesized to stimulate DCs dually via different cellular pathways. This stimulation can be sufficient by itself to provide tumor control. We used a commonly employed LNP formulation to encapsulate i) low molecular weight poly I:C for stimulation of the TLR3 pathway and ii) lipophilic cyclodextrin nanoparticles as a reservoir for small molecule payloads to affect complementary, immune stimulatory pathways inside DC. In particular, we started with CANDI to entrap a cIAP inhibitor (LCL-161) since this class of agents had previously been shown to activate myeloid cells via the non-canonical NFkB pathway. To efficiently internalize the ~38 nm CANDI nanoparticle into LNP, we modified the cyclodextrin nanoparticle surface with different lipids and found palmitic acid most efficient. We estimated that a mean LNP (120 nm) thus had ~1 CANDI nanoparticle, each with an estimated maximum capacity of ~1500 molecules of LCL-161 and ~100 molecules of poly I:C calculated using a molecular volume model. Fluorescent versions were used to determine the PK and cellular distribution. These data show that LNP17 had a T1/2 of 30 min and accumulated in the tumor, lymph nodes, and liver. These favorable PKs likely contributed to the overall efficacy of the approach.
Our approach differs from prior approaches. Poly I:C has long been used as a stand-alone therapeutic agent; unfortunately, with considerable side effects, the use of poly I:C as an immune adjuvant is more recent. Most trials use the material as an adjuvant in peptide or DC vaccination programs. Some trials have evaluated a stable form of poly I:C stabilized with poly-L-lysine and carboxymethylcellulose, Hiltonol. Additional trials have tested combinations of poly-ICLC with other immune stimulators such as granulocyte-macrophage colony-stimulating factor, resiquimod, or Montanide-ISA-51. These trials found that poly I:C and poly-ICLC effectively contribute to host anti-tumor responses as immunostimulatory components of cancer vaccines. However, many of these vaccination approaches relied upon an admixture of antigens and adjuvants. Prior direct immunotherapy studies in mouse models of melanoma used doses of poly I:C upwards of 200 µg to see anti-tumor therapeutic effects.
In contrast, our LNP-based delivery approach achieves complete tumor eradication using doses of 5 µg with a single administration, demonstrating a 40X increase in potency with our nanoformulation. This is important as currently used vaccination protocols in clinical trials are limited in their application of efficient nanoformulations to potentiate vaccine efficacy. For example, NeoVax trials in melanoma using patient-specific neoantigen peptides now rely upon the admixture of synthetic peptides with poly-ICLC. While these trials still demonstrated therapeutic responses, they suggest substantial room for improvement. More recent approaches have used intravenously administered mRNA-encoded personalized neoantigens in LNP vectors, which showed remarkable responses in difficult-to-treat pancreatic cancer.
In the current study, we show the efficacy and lack of toxicity of the hybrid LNP immunostimulant at pharmacological concentrations. LNPs may be near optimal vectors to stimulate cell-mediated immunity, inducing DCs (cDC1) as cDC1 homeostatically matures in response to apoptotic cells and typically engenders immune tolerance. LNPs themselves can trigger DC immunoregulatory maturation through cholesterol pathways, though poly I: C loaded LNPs can trigger immunogenic programs in DC. Several future experiments would be logical extensions to improve effectiveness further. For example, it is possible further to load the CANDI nanoparticle with additional small molecule payloads to induce DC3 and type 1 cell-mediated immunity, effector pathways (e.g., IL-12, CXCL9/10, IFNG). Prior research had shown that dual stimulation in macrophages via the canonical NFkB (e.g., TLR7/8 agonists such as R848) and non-canonical NFkB pathway (e.g., cIAP inhibitors) could achieve this. There is further evidence that STAT, PI3Kg, or JAK inhibition could re-program tumor myeloid cells and potentiate the effect. Second, it is possible to target the hybrid LNP to DC through antibodies (e.g., CD8A, CLEC9A, ITGAE, ITGAX, CD141, or XCR1). Such antibodies could be anchored on the surface of hybrid LNPs. Third, it is possible to arm LNP with tumor-associated antigens, including peptides or mRNA expressing tumor antigens. Irrespective of these additional modifications, the current first-generation hybrid LNP is a highly efficient dendritic cell stimulant.