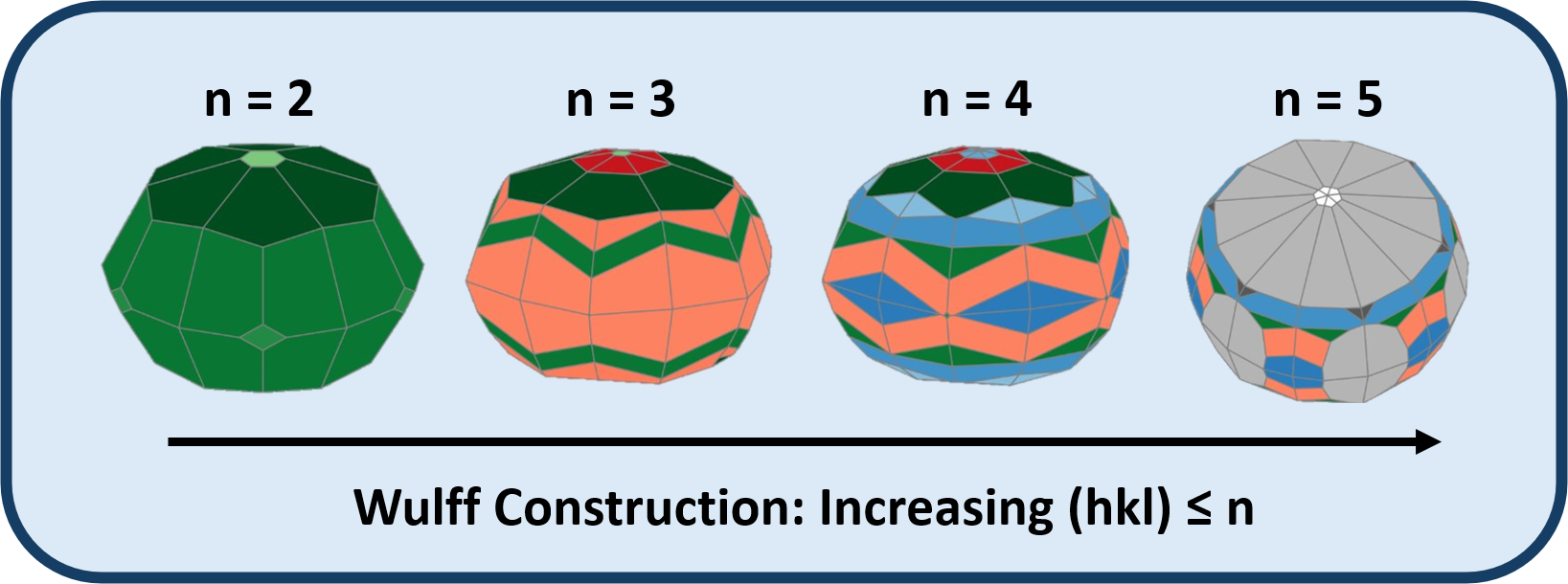
Surface energy is a critical property in understanding the stability and presence of exposed facets on the equilibrium shape of bulk crystalline materials. Surface energies can be used to develop Wulff constructions of the thermodynamically stable particle shape. As the catalytic activity of a surface is governed by the configuration of surface atoms, accurately generating these constructions is essential towards catalyst evaluation and screening. While experimental characterization techniques, such as electron backscattering diffraction (EBSD), are able to identify families of planes across a surface, they are unable to reliably identify higher index facets in polycrystalline samples. For basic metal systems, simple chemical heuristics dictate that the index magnitude of a surface is closely related to its surface energy via cleaving undercoordinated atoms. No such rule can be drawn for complex multinary intermetallic crystals where close-packed surfaces may be cleaved at higher indices. Density functional theory (DFT) enables the quantum mechanical description of these ordered materials and is able to calculate the surface energy of a Miller index given a bulk crystal system. However, the significant number of large slab models required to generate a high-index Wulff construction is too computationally intense to be deployed in a catalyst screening workflow using DFT alone. Additionally, the symmetric slab models required to calculate these energies often yield stoichiometries inconsistent with the bulk. The required stoichiometric corrections are non-trivial and can significantly influence the resulting Wulff construction.
Recent advancements in machine learning potentials (MLPs) enable rapid energy/force evaluations for systems far larger than previously accessible with DFT. Here, we present the performances of Open Catalyst Project (OCP) -based models across a low index (hkl)≤2 binary intermetallic surfaces database. MLP surface energy predictions are shown to be in strong agreement with DFT across a wide range of elemental and compositional space and result in accurate Wulff shapes. A specific intermetallic system case-study is discussed in which MLPs identify a high index facet as the most exposed surface, which is further corroborated with DFT. We also demonstrate the utility of MLPs by leveraging the models as pre-optimizers for DFT calculations, achieving an average ab inito calculation speedup of 40%. Lastly, we compare errors introduced through the reservoir identity for non-stoichiometric slab models. We introduce an intermetallic bulk formation energy-based correction to account for the stabilization caused by the heteronuclear bonds between the host and active metal species.