2025 AIChE Annual Meeting
(383al) A Generalized C++ Framework for Multiscale Kinetic Modeling in Catalytic and Electrochemical Systems
Author
My research focuses on developing multiscale modeling frameworks to understand and design catalytic and electrochemical systems under complex, dynamic operating conditions. I am particularly interested in advancing computational theory to uncover mechanistic insights in chemically and biologically complex systems, with applications spanning catalysis, electrochemistry, and energy storage. In addition, I am also interested in utilizing machine learning-based techniques to create and use models for atomic interactions and apply them towards high-throughput screening of materials for applications spanning catalyst development and drug discovery.
Abstract
Understanding and predicting reaction kinetics under dynamic and spatially heterogeneous conditions is critical for the advancement of catalytic and electrochemical technologies. The widely used mean-field microkinetic model (MF-MKM) offers a computationally inexpensive means to connect atomic-scale reaction mechanisms with reactor-scale performance through coupled rate equations. However, MF-MKM inherently neglects lateral adsorbate–adsorbate interactions and local configurational effects, which can lead to significant inaccuracies in rate predictions. Moreover, improper site accounting in MF-MKM has been shown to further compromise mechanistic accuracy, particularly in systems with non-uniform surface coverage1.
Kinetic Monte Carlo (kMC) methods provide a powerful alternative, enabling stochastic simulation of reaction kinetics with explicit spatial resolution2. By tracking individual reaction events and local atomic configurations, kMC naturally incorporates coverage effects, lateral interactions, and site heterogeneity, enhancing accuracy relative to mean-field approaches. However, this level of detail results in high computational cost, which becomes computationally intractable for multiscale or dynamically driven systems.
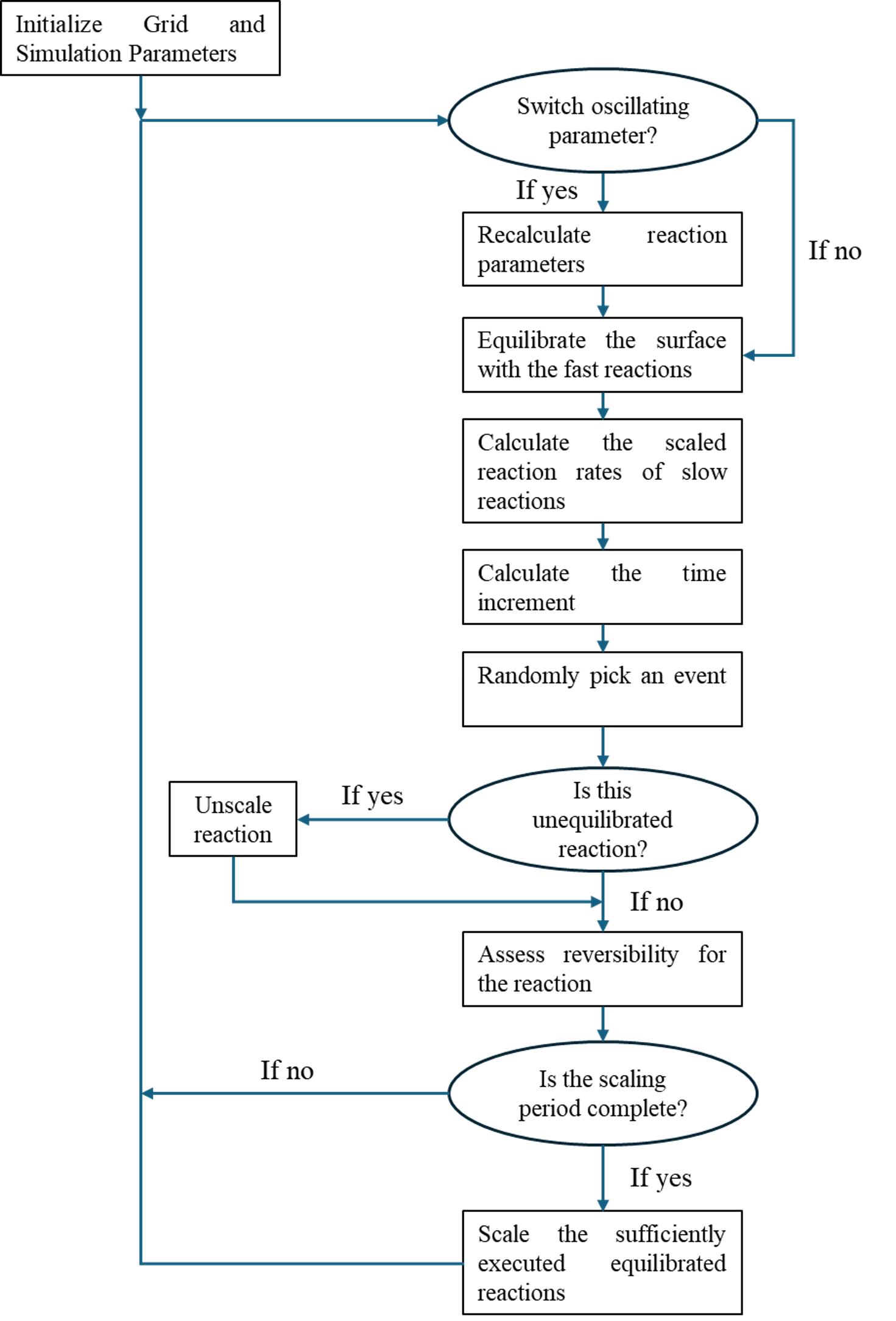
Herein, we present a generalized C++-based kMC framework designed to enable accurate yet computationally efficient simulation of complex gas–solid and electrochemical reaction networks. The codebase incorporates Neurock–Dybeck3 and Neurock–Donghai4 algorithmic acceleration techniques to group reactions at similar scales and to scale kinetic parameters for adaptive time-stepping, which significantly reduces simulation time without compromising spatial resolution. A key distinguishing feature of the framework is its ability to handle dynamically modulated external parameters such as surface charge, gas-phase partial pressures, and electrochemical potentials, allowing simulation of time-dependent or oscillatory operating conditions rarely accessible in conventional kMC tools.
The capabilities of this framework are demonstrated by applying it to ammonia synthesis on Ru surfaces under modulated surface charge conditions. Our kMC simulations, guided by quantum mechanical calculations, show that a positively charged Ru surface can enhance the ammonia synthesis rate by approximately fourfold compared to a neutral surface. Furthermore, dynamic oscillation between positive and negative surface charges leads to a ~20-fold enhancement in the rate, underscoring the catalytic benefit of non-equilibrium modulation strategies.
This framework enables multiscale kinetic modeling with unprecedented flexibility, bridging atomistic insight and process-level understanding under both steady-state and dynamic conditions. It is broadly applicable to catalytic, photocatalytic, and electrocatalytic systems where local surface dynamics play a critical role in performance.
References
1Razdan, N.K. and Bhan, A., 2021. Kinetic description of site ensembles on catalytic surfaces. Proceedings of the National Academy of Sciences, 118(8), p.e2019055118.
2Gillespie, D.T., 1977. Exact stochastic simulation of coupled chemical reactions. The journal of physical chemistry, 81(25), pp.2340-2361.
3Dybeck, E.C., Plaisance, C.P. and Neurock, M., 2017. Generalized temporal acceleration scheme for kinetic monte carlo simulations of surface catalytic processes by scaling the rates of fast reactions. Journal of chemical theory and computation, 13(4), pp.1525-1538.
4Mei, D., Hansen, E.W. and Neurock, M., 2003. Ethylene hydrogenation over bimetallic Pd/Au (111) surfaces: Application of quantum chemical results and dynamic Monte Carlo simulation. The Journal of Physical Chemistry B, 107(3), pp.798-810.