2025 AIChE Annual Meeting
(52a) Formic Acid Electro-Oxidation: Elucidating Enhancements Under Dynamic Oscillations
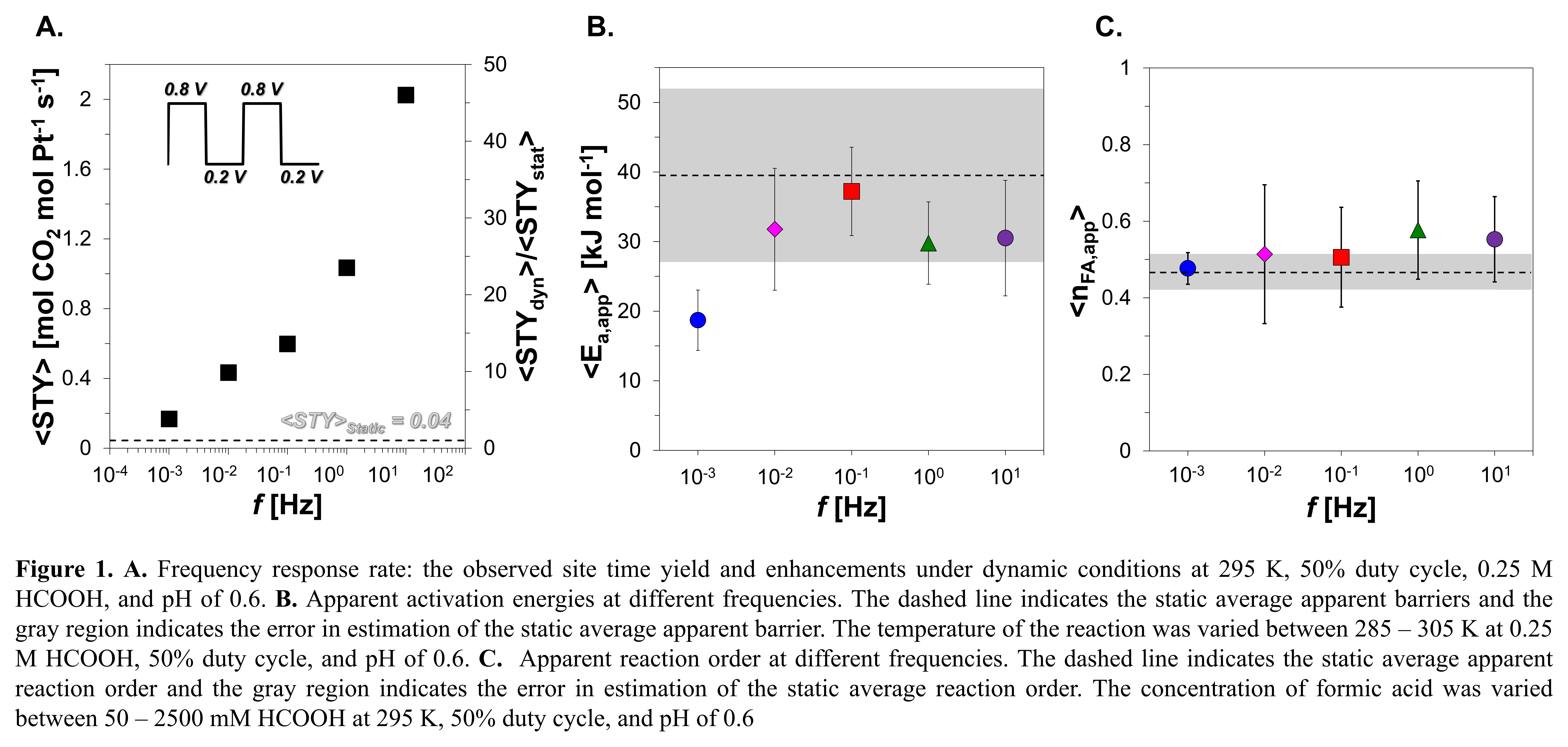
Selective acceleration of catalytic turnover within heterogeneous catalysis has been conventionally approached via material advances. The Sabatier principle depicts the limits on material properties to enhance rates, where stronger adsorbate binding leads to desorption-limiting regimes and weaker binding leads to reaction-limiting regimes. Oscillating between the distinct regimes of rate control using external energy stimuli has been shown to enhance the observed rates for formic acid electro-oxidation up to ~50x (Fig 1A). Under static conditions, apparent kinetic parameters (i.e. reaction order, activation energy) have been routinely employed to kinetically interrogate catalytic surfaces, providing quantitatively predictive and fundamentally derived rate expressions. Despite their prior utility, such kinetic interrogation approaches are yet to be employed under dynamic conditions. Here, we investigated how common kinetic parameters like apparent reaction orders and activation barriers would manifest under dynamic conditions. While dynamic catalytic turnover rate are reflected within the time domain (i.e. time average), analytical derivations of dynamic kinetic parameters were proposed to be an extent of reaction weighted-average of their respective values at each energetic state within an oscillation. To probe the hypothesis, we measured the apparent activation barriers and reaction orders for formic acid oxidation over Pt under dynamic conditions (Fig 1B, 1C), which were found to be independent of frequency and quantitatively scaled with extent of reaction weighted averages. Recognizing the scaling of apparent kinetics under oscillatory conditions by the extent of reaction at each distinct energetic state, provides greater predictive capability that helps reduce the experimental parameter space when designing a programmable catalytic surface.