2025 AIChE Annual Meeting
(253d) Estimating Water Coverage and Adsorption Isotherms at Electrolyte/Metal Interfaces Using Machine Learning Potentials
Authors
Jiawei Guo, University of California, Davis
Joel B. Varley, Lawrence Livermore National Laboratory
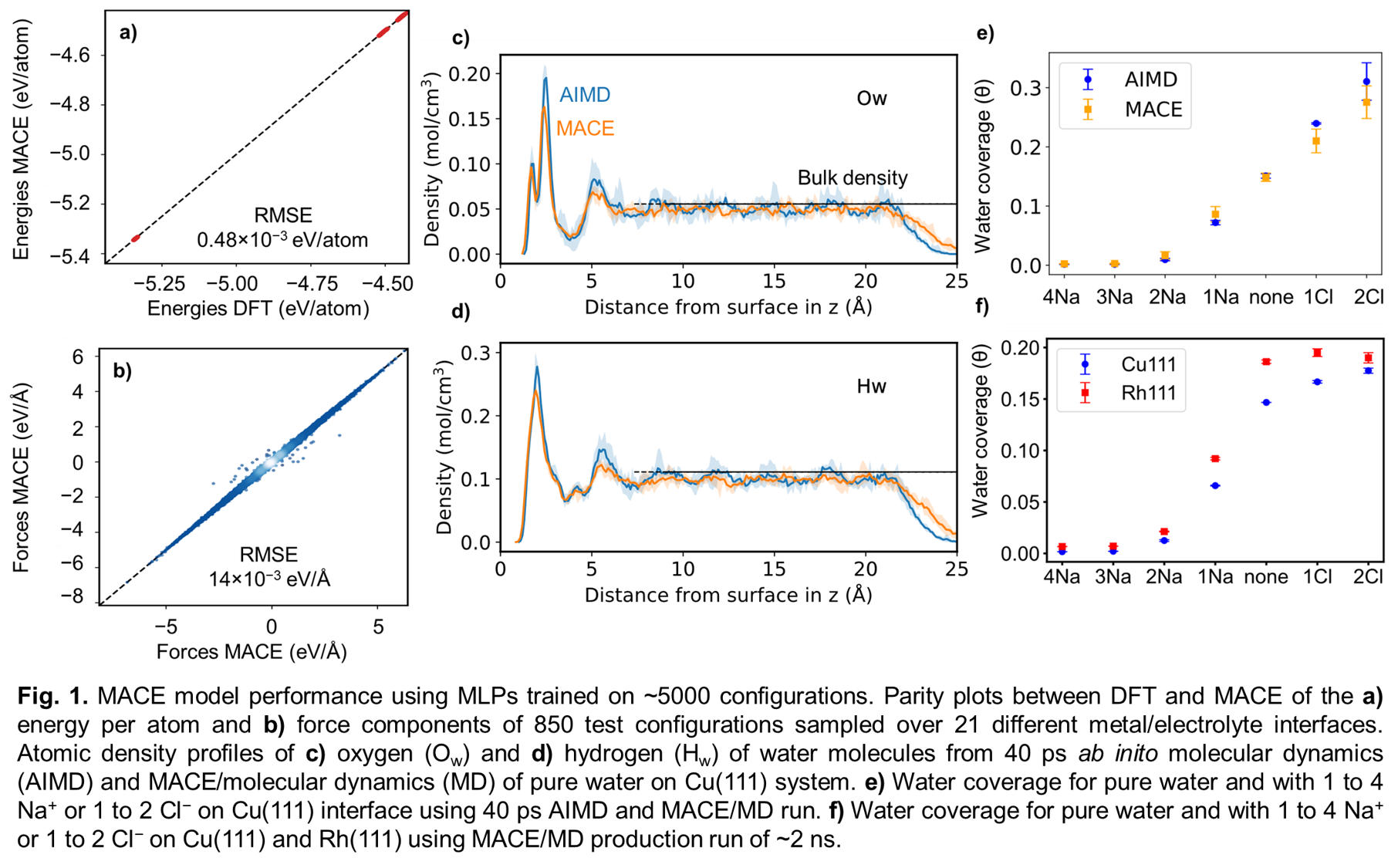
Electrocatalysis is important for many environmental and energy applications such as CO2 conversion, fuel cells, and biooil valorization. The solvent/electrode interface plays a crucial role in electrocatalytic reactions, therefore understanding the structure and properties of electrified interfaces is essential. Simulation approaches using first principles-based methods such as density functional theory (DFT) are limited in their ability to capture large length-scale and long timescale phenomena of electrified interfaces. In this work, we train machine learning based interatomic potentials (MLPs) for aqueous NaCl solutions in contact with Au, Cu, and Rh (111) electrodes. We use the MACE architecture with an active learning workflow to train the MLPs at DFT-level accuracy using ~5000 configurations for 21 different electrolyte/metal interfaces. To generate our training set, we use the revised Perdew-Burke-Ernzerhof functional with Grimme D3 long-range dispersion corrections. The MACE model achieves a root mean squared error of 0.48 meV/atom and 14 meV/Å for the energies and forces on 850 test configurations (Fig. 1 a,b), respectively. Our trained MACE model gives good agreement between ab initio molecular dynamics and MACE-based molecular dynamics (MACE/MD) simulations for several interfacial properties including the water density profiles, water orientation, and chemisorbed water coverages for different systems (Fig. 1c-e). We perform MACE/MD production runs for ~2 ns for analysis of the interfacial properties. The water coverage on Cu(111) and Rh(111) stays close to zero for more negatively charged electrodes and increases as the electrode becomes neutral or positively charged before reaching a constant water coverage at higher positive charge of electrode. Our work highlights the potential of using MLPs to perform long-scale MD simulations of electrolyte/metal interfaces, thereby enabling detailed atomistic insights into their structure and properties with high accuracy.