2025 AIChE Annual Meeting
(382av) Elucidating Enhancements for Dynamic Catalysis to Transcend Beyond State-of-the-Art
Author
Typically, metallic catalysts, particularly Pt, Pd, Cu, and Au, are used for formic acid decomposition (FAD). There exist several investigations that have innovated strategies to promote FAD selectively towards dehydrogenation. Even after developing several such strategies, it is difficult to compare and benchmark these innovations due to the variety of reaction parameters and varied ways of estimating the catalytic turnover rate. Expanding beyond FAD, catalysis research . Significant advances in synthesizing and characterizing catalysts, in combination with a detailed understanding of fundamental catalysis for estimating reaction kinetics, have informed the fundamental understanding of thousands of catalytic chemistries important to societal needs. Regardless, the ultimate goal behind all of the aforementioned endeavors is the selective acceleration of the rate of a catalytic turnover, beyond what is considered state-of-the-art. The tools and methods by which catalytic activity is assessed have been discussed in the existing literature. Conversely, how is state-of-the-art to be defined in the realm of heterogeneous catalysis? Even if defined, how can one truly transcend the limits to outperform the accepted state-of-the-art?
Coming back to FAD, the material advances, especially on Pt and Pd catalysts, involve tweaking the surface of the catalyst, which incentivizes the need to characterize the new catalytic sites. For comparisons across different materials, typically as well as specifically for benchmarking, it is critical to understand the surface identity and specifically the number of active sites participating in the reaction. For metallic catalysts, while there exist several techniques that help characterize materials, carbon monoxide (CO) chemisorption over active sites remains a commonly employed and accessible method for estimating supported metal catalyst dispersion. Measurements of catalyst dispersion through chemisorption are typically performed using commercially available instruments using static or dynamic chemisorption techniques, with comparable results between the two. Despite the relative ease of the approach, commercial instruments are expensive and thus could benefit from cheaper alternatives. An alternative technique that is relatively inexpensive, reproducible, and still ensures broad limits of detection would greatly enhance the estimation of metallic dispersion.
Using electrochemical overpotentials as driving forces for FAD, via formic acid electrooxidation (FAEO), is also critical to discuss, given the huge potential as a fuel in DFAFCs. Within FAEO, material advances have shown to be a key strategy in advancing heterogeneous catalysis, whose limitations are best depicted by the Sabatier volcano. Within the Sabatier limits, weakly bound regimes favor desorption over surface reaction, whereas strongly bound regimes favor the surface reaction over desorption. Conventionally, the design of catalysts has been directed towards fabricating materials that show intermediate binding energies, closer to the peak of Sabatier’s volcano, acting as our state-of-the-art. A key strategy involving the modulations of external energy stimuli, i.e., dynamic catalysis, was employed to break free of such Sabatier limitations and hence the state-of-the-art. Dynamic catalysis, as a strategy, entails pulsing the external energetic stimuli to swing between distinct, well-defined adsorption regimes periodically. Even though enhancement due to dynamic catalysis is an intriguing field of scientific research that has prompted several investigations, the chemical sensitivities to the rate under dynamic conditions are still an unexplored avenue.
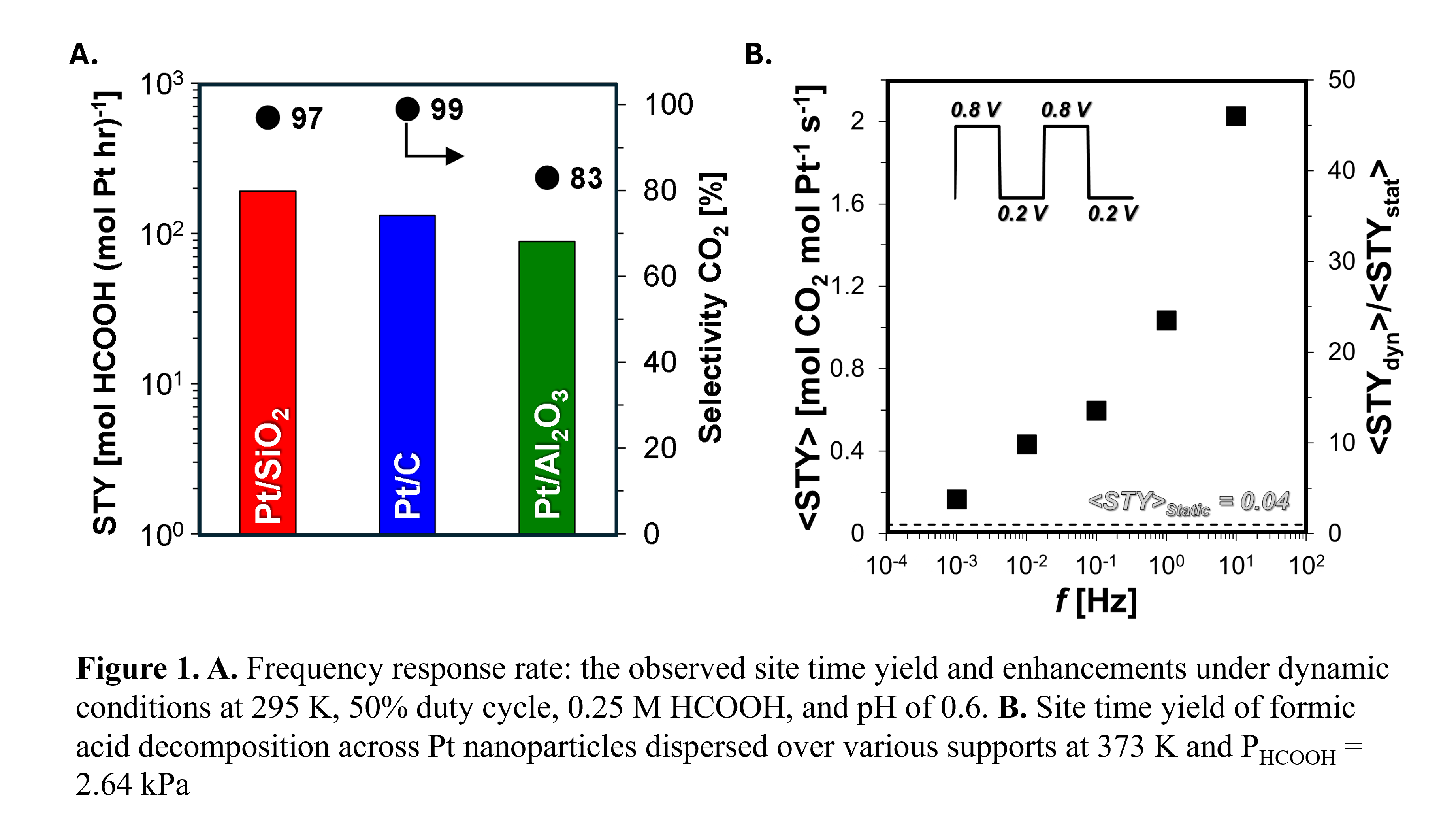
Through the series of investigations, we shall discuss first the state of the art in heterogeneous catalysis and, specifically, the development of a benchmarking database for experimental catalytic values, CatTestHub. As a tool, CatTestHub (cpec.umn.edu/cattesthub) seeks to standardize data reporting across heterogeneous catalysis, providing an open-access community platform. Here, we highlight using the database to develop structure-function relations for distinct chemistries. The selectivity to the formic-acid dehydration pathway over alumina-supported Pt nanoparticle (17%) is significantly distinct from that across carbon and silica supports (1%), despite the insensitive site time yield, indicating the presence of support interactions (Figure 1A). The database enables such discussions of insights that form the foundations for probing more complex problems. CatTestHub is a valuable tool for chemical catalysis research, enabling a more community-wide benchmarking effort. The growth of the database promises to enable modeling based on carefully curated data and improve the precision of experimental investigations, ultimately expediting the discovery of efficient catalytic materials.
To bolster the data highlighted in CatTestHub, we also delineate an alternative chromatographic chemisorption method that helps determine the metal dispersion and, hence, the active site density. The alternative technique helps in the instantaneous in-situ estimation of the total number of active sites. The newer technique is benchmarked against a known standard for estimating the number of active sites to validate its accuracy.
With the state-of-the-art, well-defined, we explore the avenues of dynamic modulation of overpotentials to drive catalytic turnover for FAEO. Oscillating between the distinct regimes of rate control using external energy stimuli has been shown to enhance the observed rates for FAEO up to ~50x (Figure 1B). Under static conditions, apparent kinetic parameters (i.e. reaction order, activation energy) have been routinely employed to kinetically interrogate catalytic surfaces, providing quantitatively predictive and fundamentally derived rate expressions. Despite their prior utility, such kinetic interrogation approaches are yet to be employed under dynamic conditions. Here, we investigate how common kinetic parameters like apparent reaction orders and activation barriers would manifest under dynamic conditions. While dynamic catalytic turnover rate are reflected within the time domain (i.e. time average), analytical derivations of dynamic kinetic parameters are shown to be an extent of reaction weighted-average of their respective values at each energetic state within an oscillation. To probe the hypothesis, we measure the apparent activation barriers and reaction orders for formic acid oxidation over Pt under dynamic conditions, which is found to be independent of frequency and quantitatively scaled with extent of reaction weighted averages. Recognizing the scaling of apparent kinetics under oscillatory conditions by the extent of reaction at each distinct energetic state, provides greater predictive capability that helps reduce the experimental parameter space when designing a programmable catalytic surface.
Throughout the series of investigations presented here, we aim to tackle broader perspectives and, due to its high applicability in closing the circular hydrogen economy, investigate FAD over metals as the model reaction chemistry to define our sandbox. The investigations into the state-of-the-art and fundamentally elucidating dynamic catalysis serve to help the field of heterogeneous catalysis grow.
Research Interests:
My research integrates core principles of catalysis, reaction engineering, and advanced materials science to develop innovative solutions to complex chemical engineering challenges. By combining computational modeling (such as ab initio DFT simulations) with rigorous experimental work — including reaction kinetics in packed bed reactors and electrocatalytic surface investigations — I have built a strong foundation in both theoretical and applied research.
As I transition to industry, I am especially interested in contributing to the pharmaceutical, semiconductor, and oil and gas sectors, where precision, scalability, and innovation are vital. Specifically, I aim to:
- Gain experience and expertise in advanced materials and surface characterization techniques (e.g., TEM, SEM, XPS), enabling me to optimize catalyst surfaces for energy and refining applications, fine-tune pharmaceutical formulations, or enhance semiconductor materials at the nanoscale.
- Apply my background in solid-state physics and reaction engineering to support large-scale process design and scale-up — from pharmaceutical manufacturing and semiconductor fabrication to refining and petrochemical operations — ensuring high efficiency, safety, and product quality.
- Leverage data science and advanced analytics to drive process optimization, predictive maintenance, and continuous improvement initiatives, aligning with modern industry goals for operational excellence and sustainability.
Overall, I am driven by a passion for solving high-impact, interdisciplinary problems and translating fundamental scientific insights into scalable, real-world solutions. My goal is to contribute to advancing products and processes that enhance performance, safety, and sustainability across these critical industries.