2025 AIChE Annual Meeting
(382p) Electrochemical CO2 Conversion to Ethylene and Ethanol with High Faradaic Efficiency, Lower Cell Potential and Electrolyte Recycling
I am actively involved in the frontiers of electrochemistry research through my work on the electrochemical reduction of CO₂ to high-value C₂+ products, with a strong focus on improving selectivity, catalyst stability, and system scalability. This project is funded by two major industries (Saudi Aramco and Braskem USA).
The electrochemical reduction of carbon dioxide (eCO₂R) to multi-carbon (C₂⁺) products such as ethylene and ethanol holds immense promise for a sustainable alternative to traditional crude oil and support global initiatives like the U.S. Department of Energy’s Clean Fuels and Products Shot prioritizing the development of low-carbon fuels to achieve a net-zero carbon future. Among various pathways, bicarbonate electrolysis emerges as an attractive alternative to gas-phase CO₂ reduction using gas diffusion electrodes (GDEs), as it allows the direct use of CO₂ capture solutions without requiring energy-intensive regeneration, purification, or compression of CO₂ gas. However, several critical bottlenecks limit the practical implementation of bicarbonate-fed CO₂ electrolysis systems, particularly when targeting selective and energy-efficient production of C₂⁺ products. The key challenges include: (1) Local pH Rise and CO₂ Depletion: The consumption of protons and generation of hydroxide ions (OH⁻) during CO₂ electroreduction causes a sharp increase in local pH, which shifts the carbonate equilibrium toward bicarbonate (HCO₃⁻) and carbonate (CO₃²⁻), thereby reducing the availability of dissolved CO₂ near the catalyst surface.(2) High Overpotential: The conversion of HCO₃⁻ to CO₂ is thermodynamically uphill, requiring additional energy (ΔE ≈ 0.21 V vs SHE), which results in higher applied cell potentials compared to gas-fed systems. (3) Mass Transport Limitations: The elevated pH at high current densities reduces CO₂ diffusion flux toward the active sites, diminishing conversion efficiency. (4) Loss of Active Cu⁺ Species: Prolonged operation under reducing conditions leads to the reduction of catalytically beneficial Cu⁺ and Cu²⁺ species to metallic Cu⁰, which reduces selectivity for C₂⁺ products and promotes the hydrogen evolution reaction (HER). (5) Dynamic Electrolyte Evolution: Accumulation of liquid products such as ethanol, acetic acid, or formic acid during operation alters the electrolyte composition, local viscosity, and pH buffering behavior, which in turn affects long-term selectivity and stability.
To address these complex and interrelated limitations, we have developed and validated a comprehensive electrochemical platform integrating four novel strategies:
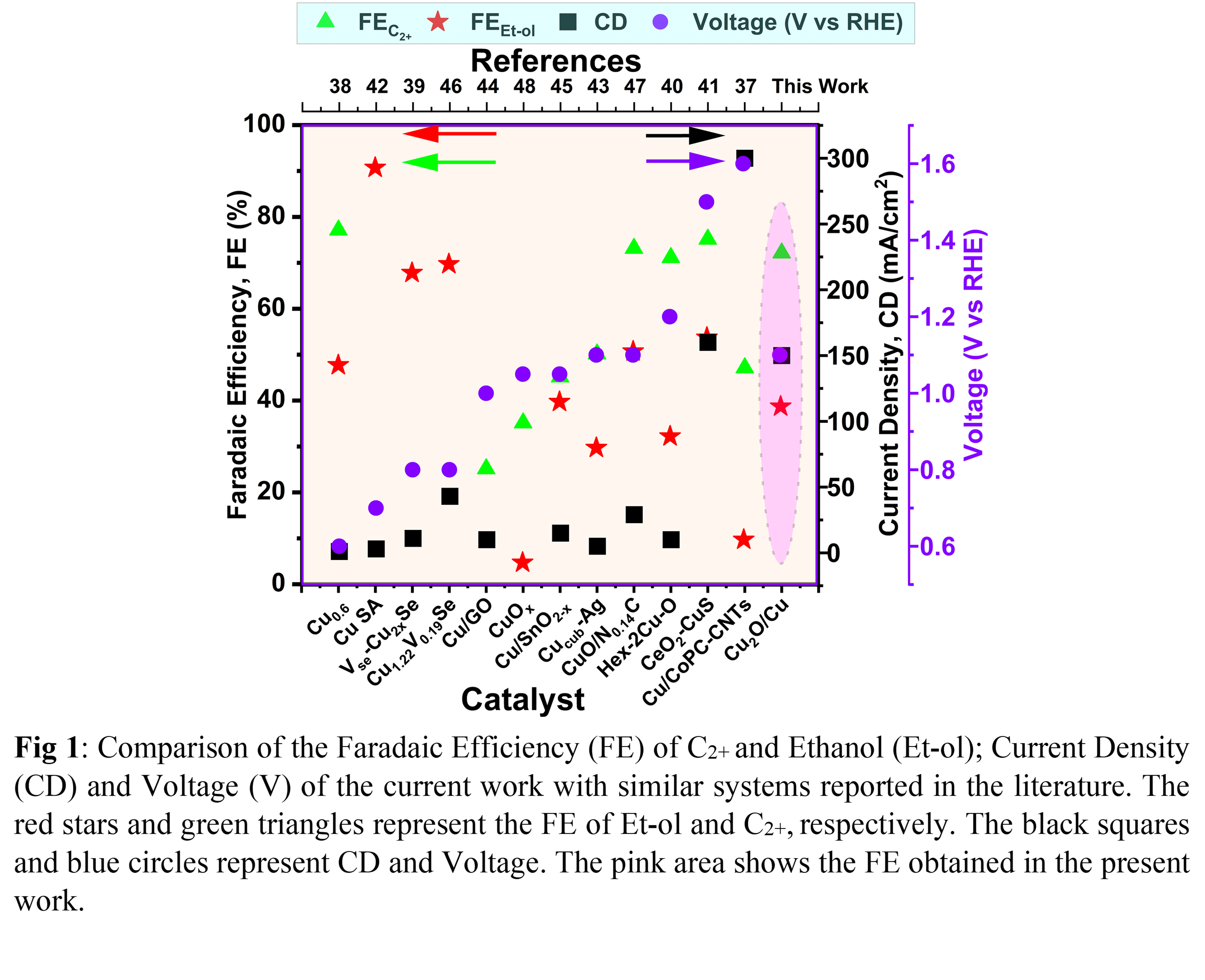
- In-situ Catalyst Activation via Pulsed Electrolysis: A pulsed electrolysis strategy is designed to dynamically modulate the catalyst state and the reaction microenvironment, addressing the dual challenges of catalyst deactivation and local CO₂ depletion. Using a Cu₂O/Cu mesh pre-catalyst, we implemented a multi-segment pulsed waveform (cycle time t = 4 s), where short oxidative pulses restore Cu⁺ and Cu²⁺ states, and reductive segments initiate CO₂ reduction under freshly regenerated surfaces. Under optimized conditions, we achieved 72% Faradaic efficiency (FE) for C₂⁺ products, 52% FE for total liquid products, and 39% FE for ethanol at a current density of 150 mA/cm² and –1.1 V vs RHE.
- Multiscale-Multiphysics Modeling of Dynamic pH and Surface Species: To elucidate the underlying mechanisms of enhanced C₂⁺ formation under pulsed operation, we developed a comprehensive multiscale, Multiphysics model that simulates the dynamic behavior of surface copper species and local electrolyte composition. The model integrates: Time-dependent potential waveforms, Surface microkinetic modeling of Cu⁰, Cu⁺, and Cu²⁺ species, Mass transport and buffering of CO₂, HCO₃⁻, CO₃²⁻, and OH⁻, and Experimental Faradaic efficiencies as inputs to constrain kinetic rate constants. The alternating redox segments during pulsed electrolysis lead to pH oscillations between ~3.5 and 9.3, which buffer local conditions, allowing CO₂ to regenerate to ~34 mM and preventing irreversible equilibrium shifts toward CO₃²⁻. A buffering delay (~4.0 to 4.2 s) during the oxidative phase is attributed to the buffering action of bicarbonate, which helps modulate sharp pH transitions. This predictive model offers valuable design guidelines for pulse durations and amplitudes to optimize surface states and electrolyte behaviour in real time, and it serves as a framework for reactor and process scale-up.
- Halide-Mediated Electrochemical Engineering: We next investigated the role of halide ions (Cl⁻, Br⁻, I⁻) as electrolyte additives to stabilize Cu⁺ species, suppress HER, and reduce cell voltage through increased ionic conductivity and surface restructuring. Through systematic variation of halide concentrations (0.1 M–3 M) and cathodic current densities (50–300 mA/cm²), we identified iodide (I⁻) as the most effective. When combined with pulsed electrolysis, this strategy yielded: 75% FE for C₂⁺ products, 60% for total liquid products, and 39% ethanol at 250 mA/cm² and –1.1 V vs RHE, while maintaining low full-cell potentials relative to halide-free systems. Operando techniques, including in-situ Raman and XRD, verified halide-induced stabilization of Cu(I) oxidation states and modified surface morphology. The synergy between halide-driven surface engineering and pulse-induced dynamics leads to a self-regulated reaction environment that facilitates C–C coupling over HER. Importantly, halides not only act as electronic structure modulators but also enhance ionic transport, thereby reducing internal resistance and improving energy efficiency—a critical requirement for scale-up and integration into CO₂ capture systems.
- Electrolyte Recycling Informed by Byproduct Accumulation: While catalyst development and reaction engineering are often emphasized, the dynamic evolution of electrolyte composition is a critical, yet underexplored, factor in long-term electrochemical CO₂ reduction. Our study examined the influence of representative byproducts: formic acid, acetic acid, ethanol, methanol, propanol, and ethylene glycol on pH buffering capacity, electrolyte viscosity and conductivity, competitive adsorption on active sites, and overall system durability. We found that ethanol and acetic acid modulate local pH, improving C₂⁺ selectivity in the short term. However, accumulation of alcohol and organics leads to higher viscosity, lower diffusivity, and non-ideal solvation effects, impacting conversion rates and catalyst accessibility. Some compounds also interfere with CO₂ adsorption or block active sites, reducing efficiency. Periodic or continuous electrolyte recycling via membrane separation or flow exchange improved system performance, restoring conductivity and pH buffering capacity. This underscores the importance of treating electrolyte as a dynamic component of the reactor, with feedback loops between reaction products and operating conditions.
- Toward Scalable CO₂-to-Fuel Technologies: By integrating pulsed electrolysis, predictive modeling, halide-assisted engineering, and electrolyte management, we demonstrate a robust strategy to overcome the fundamental limitations of bicarbonate-fed CO₂ electrolysis. This unified platform achieves: High C₂⁺ selectivity (up to 75%), Lower cell potentials (–1.1 V vs RHE), Current densities >250 mA/cm², and Stable operation through dynamic catalyst and electrolyte control.
The combined approach bridges the gap between lab-scale innovation and industrial implementation, offering a blueprint for the development of modular, energy-efficient, and scalable electrochemical reactors that support clean fuels and carbon-negative goals. Our findings contribute to the growing ecosystem of CO₂ utilization technologies and align with the U.S. Department of Energy’s mission to develop integrated platforms for carbon capture, conversion, and utilization (CCU).