2025 AIChE Annual Meeting
(526e) Electrochemical C-N Bond Formation: A Density Functional Theory Study of Urea Synthesis on Cu and Cu Alloys
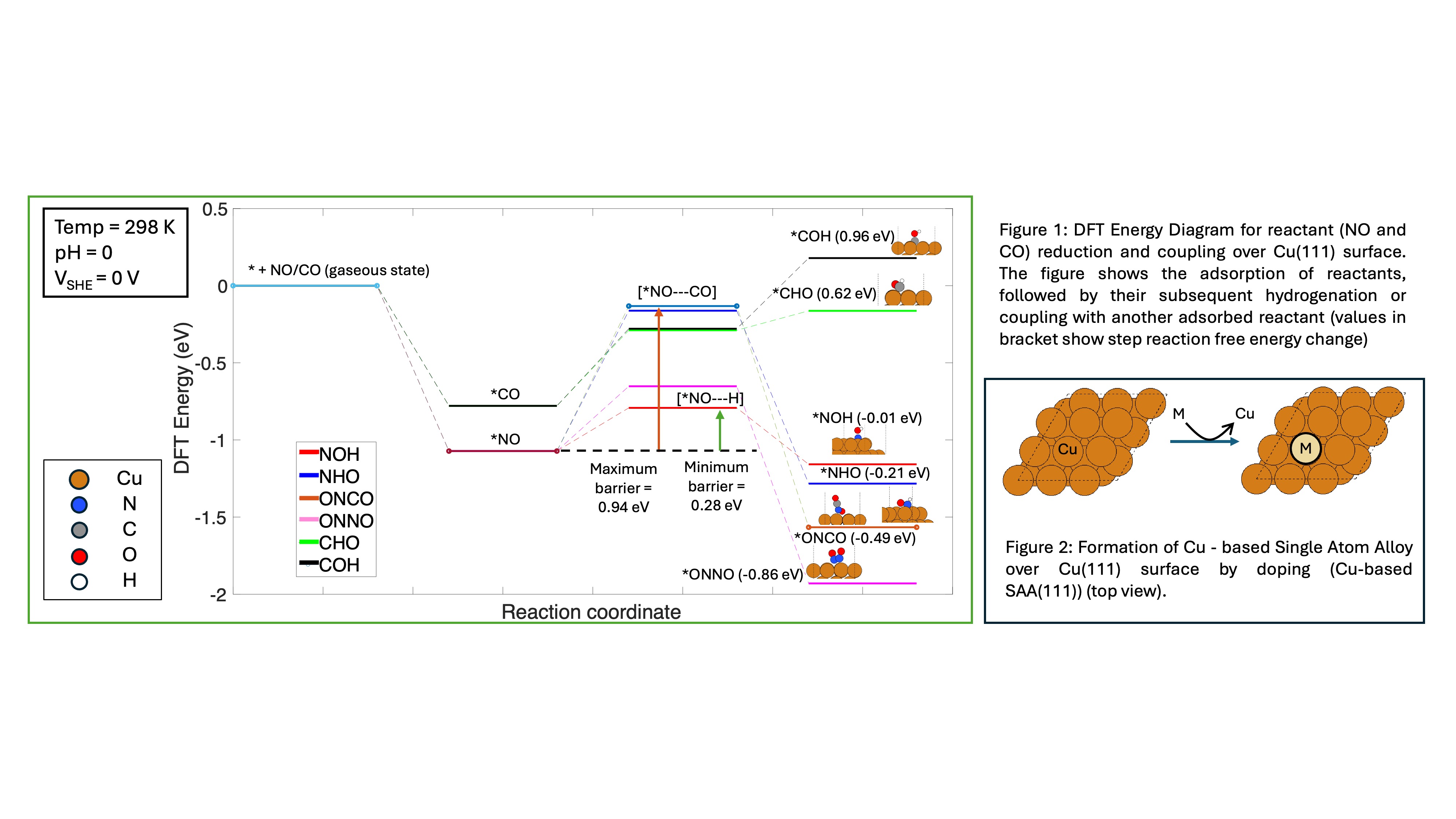
In this work, we combine Periodic Density Functional Theory (DFT) calculations with electrokinetic analyses to elucidate the mechanism of the urea synthesis in an aqueous electrochemical environment. Motivated by reports of electrocatalytic reduction of small molecules in the literature, we begin our analysis on single crystal Cu(111) and Cu(211) surfaces, as urea synthesis has been observed at ~-0.75V vs. SHE on Cu-loaded gas diffusion electrode. We propose a comprehensive mechanism involving adsorbate hydrogenation and coupling through multiple reaction pathways, ultimately leading to urea and non-selective products. The energetic parameters associated with corresponding elementary steps are obtained from DFT, and voltage dependence of the proton-coupled electron transfer steps is determined using Nernstian thermodynamics. Initial NO and CO hydrogenation and their coupling over catalytic surfaces are found likely to be rate-limiting reactions (Figure 1), as subsequent steps are thermodynamically spontaneous. Further, we analyze the changes in reactant binding energies, as well as associated changes in reaction mechanisms, that occur when copper surfaces are doped with various elements to form single atom alloys (Figure 2). We assess the impact of these alloys on product selectivity, and predict which such alloys will be stable under aqueous acidic conditions. We close by outlining future challenges that must be overcome to realize practical synthesis of urea under electrochemical conditions.