2025 AIChE Annual Meeting
(197b) Direct Propylene Epoxidation By Water Oxidation on PdPtOx Electrocatalysts
Authors
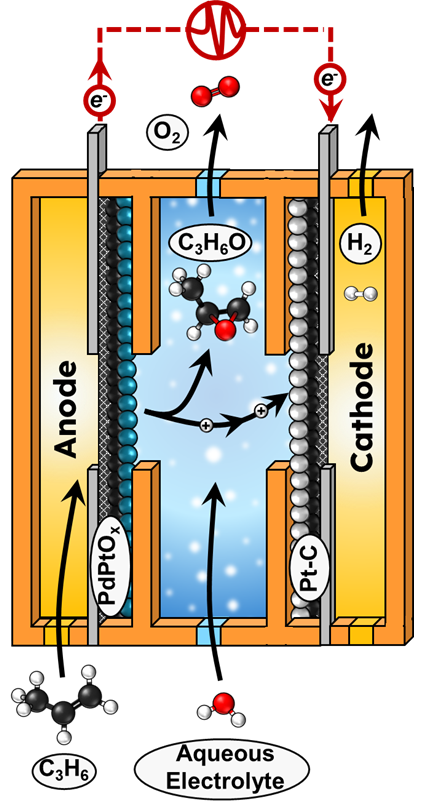
Epoxidation rates increase with H2O concentration with a 2nd-order dependence, suggesting two water molecules participate in the kinetically relevant step (KRS), consistent with (H2O/D2O) kinetic isotope effects. Epoxidation rates increase sublinearly with C3H6 pressure (n=0.4-0.7), consistent with C3H6* species saturating the surface. Rates and FEs of epoxidation increase exponentially with potential in aqueous electrolytes with Tafel slopes of 102 mV dec-1, consistent with one-electron transfer to the most abundant reactive intermediate in the KRS. These findings agree with a mechanism involving the adsorption of H2O and C3H6 to the surface of PtO active sites, which oxidize by sequential proton-coupled electron transfer steps (OH*→O*→OOH*→O2). The KRS, however, likely involves the oxidation of O* to OOH* by two H2O molecules, in which the fate of OOH* determines the selectivity of forming C3H6O or O2 by distinct charge transfer steps. Overall, we elucidate the activate phase of PdPtOx and determine the reaction mechanisms of epoxidation, leading to record epoxidation rates and FEs.