2025 AIChE Annual Meeting
(376b) Direct Partial Oxidation of Methane to Methanol at Plasma-Catalyst-Liquid Interfaces
Electrified approaches for the partial oxidation of methane by nonthermal plasmas are a promising platform to help overcome these challenges. Nonthermal plasmas are characterized by electron temperatures much higher than the ions or neutral molecules within the plasma. Unlike thermal processes, nonthermal plasma activation enables non-equilibrium chemistry, allowing reactions to proceed at lower bulk temperatures but with high-energy local interactions, which is beneficial for activating the strong C-H bond in methane yet inhibiting methanol overoxidation. Plasma-driven solution electrochemistry is an emerging frontier where highly energetic plasmas are interfaced with liquid environments to exploit unique oxidative and reductive chemical pathways. In particular, at aqueous interfaces the production of •OH from plasma-activated water has garnered attention for its simple and cost-effective application in oxidative chemistries. Combining the unique reactive environments at plasma-liquid interfaces with heterogeneous catalysts have been seldom reported, but up to this point has been achieved by dispersing catalyst into the liquid, thereby minimizing catalyst-plasma interactions due to mass transport limitations and short lifetimes of plasma species.
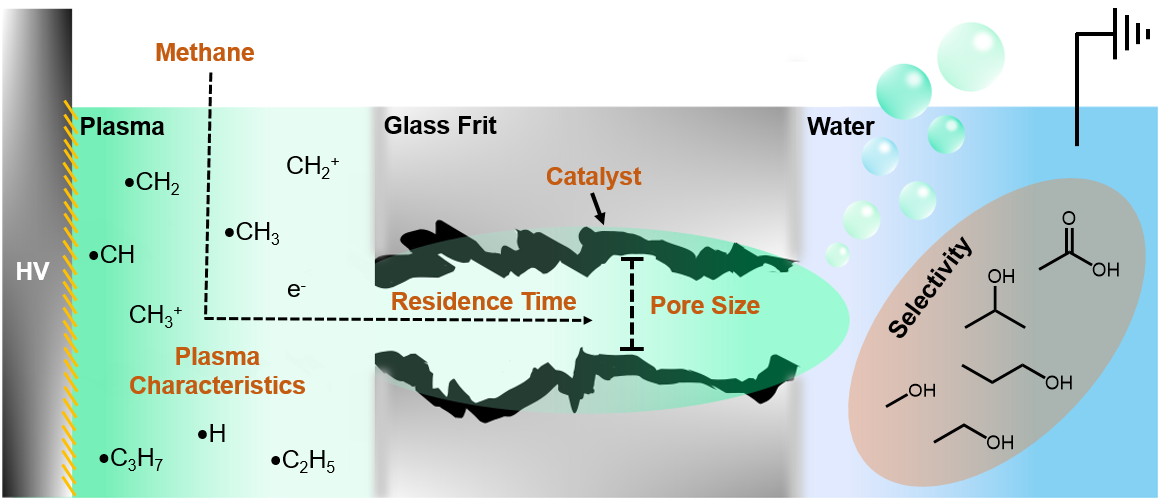
Herein, we introduce an approach to overcome several key limitations for direct partial methane oxidation by combining foundational concepts from heterogeneous catalysis with those of plasma-driven solution electrochemistry. In this work, we present a novel plasma reactor design that: (1) leverages the non-equilibrium dynamics of non-thermal plasmas to activate kinetically sluggish methane and water at low temperatures, (2) utilizes in situ generated oxidants as alternatives to molecular oxygen, (3) integrates a catalyst to enhance reaction selectivity through increased interactions with reactive plasma species, and (4) rapidly partitions produced methanol into liquid to quench its overoxidation. To achieve this, we embed CuO catalysts directly into the pores of commercially available fritted-glass gas dispersion tubes where methane plasma is discharged and submerged underwater, creating the plasma-catalyst-liquid interface. We investigate key experimental variables—including membrane pore size, residence time, catalyst loading, and plasma electron energy distribution function—to optimize the plasma-catalyst-liquid interface (PCLI) system for high methanol liquid selectivity.
From simulations and experimental data, we illustrate a mechanistic picture of the complex dynamics within the reaction environment that drives improved methanol production and selectivity. Plasma modelling suggests that vibrationally hot methane are the dominant species at the PCLI under relevant experimental conditions. Systematic experimental optimization reveals that proximity-driven interactions between the plasma and CuO catalyst are crucial for achieving high methanol selectivity. Frit porosity influences plasma-catalyst-liquid interactions as decreasing the pore size increases the number of pore channels and catalytic surface area, increasing the number of interactions between catalyst and plasma-species by a proximity effect. Conversely, the decreasing pore size limits electron density in the channels, reducing the frequency of plasma-liquid interactions occurring at this interface. Along mass transport consideration, we show that the plasma residence time at the PCLI must be balanced to optimize catalyst interactions and quenching rates of methanol to achieve high selectivity. The role of the plasma electron energy distribution function is also investigated. Increasing applied plasma voltage, while increasing liquid production rate, also decreases the selectivity of methanol due to overactivation of methanol, leading to its decomposition pathway to carbonyl products. Improvements to methanol selectivity can be made by inhibiting the production of C2+ radicals in the plasma by introducing argon gas. Argon acts as a dilutant to inhibit methyl-methyl radical recombination and by further activating methane via Penning ionization.
By localizing the relevant chemistry within the plasma-catalyst-liquid interface, we achieve exceptional performance with minimal catalyst loading (0.12 mgCuO/mLH2O ) and high liquid selectivity for methanol (>97%)—a result unattainable with conventional catalyst dispersion in liquid. This work establishes a scalable, electrified oxidation platform for methane conversion with broad implications for plasma-driven solution electrochemistry such as catalytic oxidation reactions of higher order alkanes, phenol, and other complex organic oxidation reactions.