2025 AIChE Annual Meeting
(705c) From DFT to Mlps: Advancing Surface Adsorbate Diffusion Simulations with Active Learning
Authors
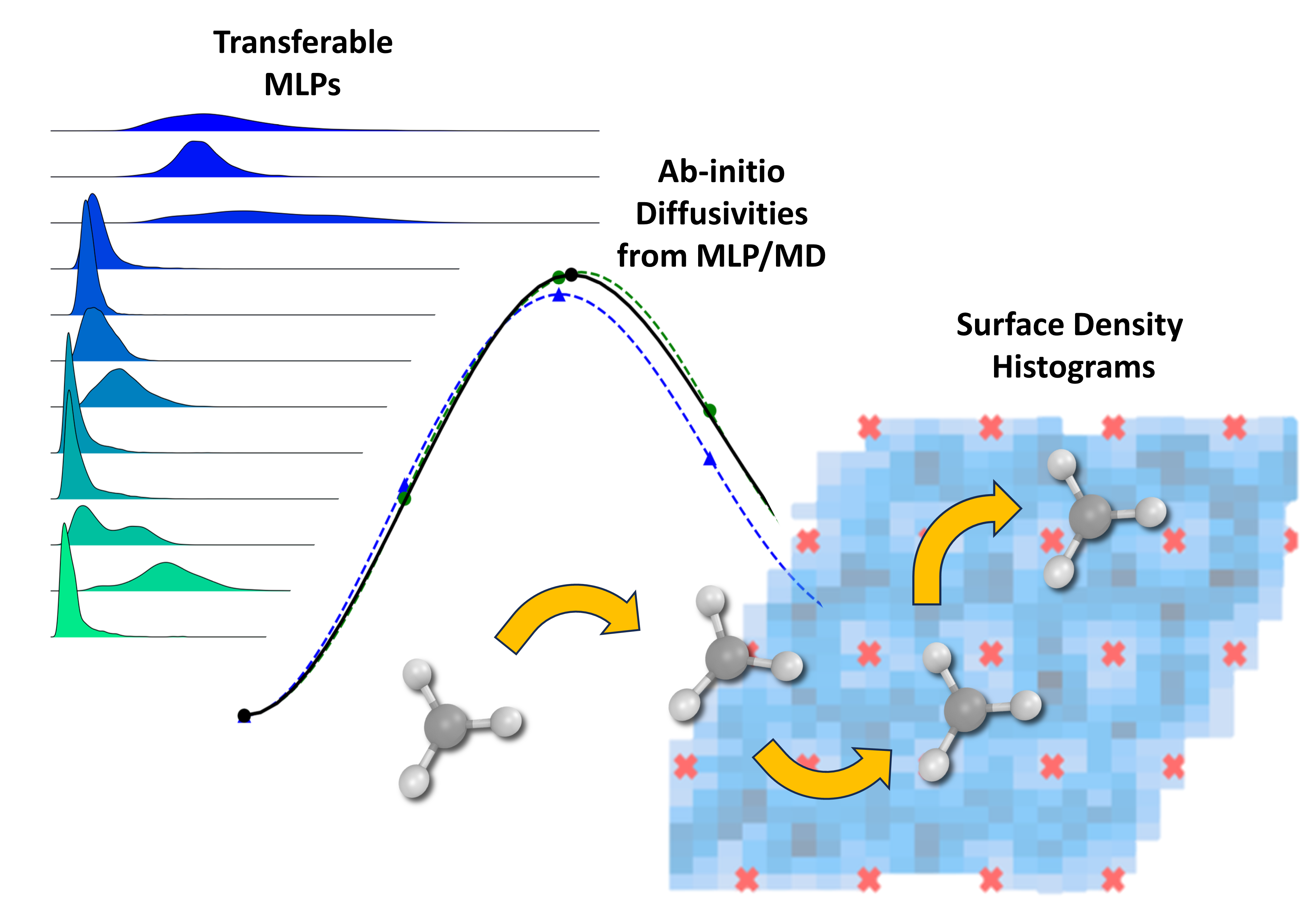
We apply the resulting MLP to perform long-timescale molecular dynamics (MD) simulations of adsorbate diffusion on a Ag(111) surface. These simulations allow for the extraction of temperature-dependent diffusivities under realistic thermal conditions, capturing anharmonic effects and rare events that are typically inaccessible using static DFT calculations. Analysis of the MLP-MD trajectories reveals notable deviations from diffusion barriers estimated using the nudged elastic band (NEB) method, which often relies on idealized transition pathways and may neglect entropic contributions. Our findings highlight the limitations of NEB-based approaches for accurately characterizing surface diffusion, particularly in systems with complex energy landscapes or multiple hopping mechanisms. In contrast, the MLP-MD approach provides a more comprehensive view of dynamic behavior by naturally incorporating thermal fluctuations and statistically relevant pathways.
While the present study focuses on prototypical adsorbates on Ag(111), the training workflow and model development strategy are system-agnostic and readily extendable to other surfaces, adsorbates, and reaction environments. This work demonstrates the power of combining active learning with machine learning potentials to bridge the accuracy of DFT with the sampling efficiency of classical MD, paving the way for predictive modeling of surface kinetics and transport processes in heterogeneous catalysis and related domains.