2025 AIChE Annual Meeting
(320a) DFT Investigation of Subsurface Metal-Oxygen Ligand Influences on Adsorption Energetics in IrO2-RuO2 Heterostructured Monolayers
Authors
Suriya Ramasubramian, University of Florida
Christopher Lee, University of Florida
Alvaro Loaiza, Louisiana State University
Randall Meyer, Exxonmobil
Craig Plaisance, Louisiana State University
David Hibbitts, University of Florida
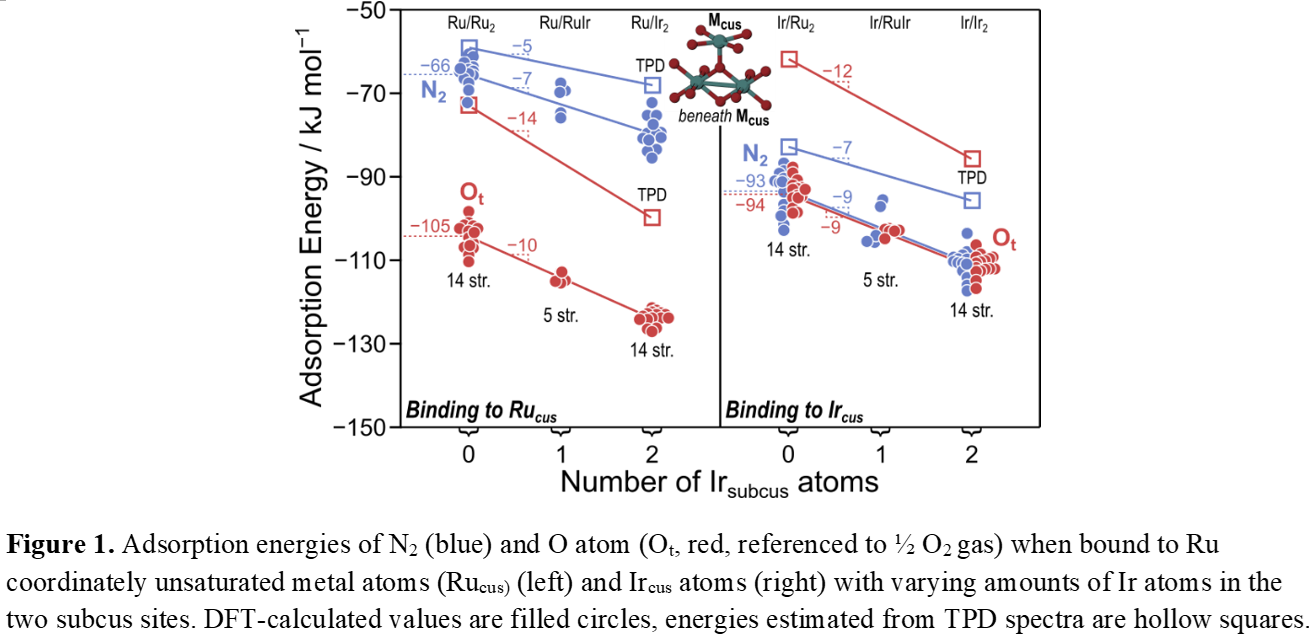
Rutile oxides IrO2 and RuO2 are important catalysts for oxygen evolution and alkane oxidation. Using density functional theory (DFT) and temperature programmed desorption (TPD), we investigate binding properties of IrO2-RuO2 heterostructures to advance catalyst design. TPD measurements reveal N2 binds 13 kJ mol−1 more weakly on single-layer IrO2/RuO2 compared to multi-layer IrO2, while O atoms show 24 kJ mol−1 weaker binding. Conversely, on single-layer RuO2/IrO2, both N2 and O atoms bind stronger (by 9 and 27 kJ mol−1) than on multi-layer RuO2, demonstrating significant subsurface influence. DFT calculations confirm these trends, showing N2 binding strength follows: IrO2 (−111 kJ mol−1) > 1L-IrO2/RuO2 (−94 kJ mol−1) > 1L-RuO2/IrO2 (−81 kJ mol−1) > RuO2 (−62 kJ mol−1), while O atoms follow: 1L-RuO2/IrO2 (−124 kJ mol−1) > IrO2 (−109 kJ mol−1) > RuO2 (−103 kJ mol−1) > 1L-IrO2/RuO2 (−93 kJ mol−1). Studies of atomically mixed surfaces reveal binding energetics are primarily determined by the surface metal site and subsurface metals directly beneath it, with each Ir atom in subsurface positions strengthening binding by 8-10 kJ mol−1. Our computational framework based on alchemy and entanglement decomposition shows when adsorbates bind, electron density shifts toward subsurface oxygen atoms. And thus, Replacing Ru with Ir at subsurface positions introduces higher positive charge that stabilizes this electron redistribution, strengthening binding. Notably, subsurface Ir enhances binding for both N2 and O atoms, while surface metal sites have opposite effects—enhancing N2 binding but weakening O binding. This research provides insights into engineering rutile oxide heterostructures to tune catalytic properties and offers a unified approach to understanding site and ligand effects in metal oxide catalysts.