2025 AIChE Annual Meeting
(389at) Designing Anti-Amyloidogenic Agents with Generative AI and Molecular Modeling
Authors
Alzheimer’s disease is a progressive neurodegenerative disorder. It is characterized by the buildup of amyloid-beta (Aβ) plaques in the brain, which results in neuronal impairment and cognitive decline. Targeting this pathological hallmark, anti-amyloidogenic agents have gained attention as potential therapeutics by preventing Aβ formation, blocking its aggregation, and enhancing its clearance. However, the discovery of effective anti-amyloidogenic agents remains a slow and resource-intensive process, heavily dependent on translational research. Furthermore, the detailed mechanisms, particularly the structural and thermodynamic aspects of how these agents inhibit Aβ42 aggregation, are still unclear. To address these challenges, integrating AI/machine learning with molecular modeling offers a powerful strategy for drug discovery. In this approach, reinforcement learning, such as recurrent neural networks (RNNs) and transformers, was employed for the rapid AI-generation of novel molecules, and molecular dynamics simulations was utilized for investigating the molecular mechanisms underlying the anti-aggregation effects on Aβ42.
Method:
Using an active learning iteration-based approach,1,2 as a cycle of designing a new molecule, a dataset of thirty anti-amyloidogenic molecules was compiled from our previous experimental studies3,4 and the CHEMBL database. The molecular structures in this dataset were converted into Simplified Molecular Input Line Entry System (SMILES) format using OpenBabel for subsequent analysis. Then, new compounds were generated through the Reinvent reinforcement learning framework.5 In this process, binding free energy calculations were carried out using the Molecular Mechanics/Poisson–Boltzmann Surface Area (MMPBSA) method, while ChemProp, a directed message-passing neural network (D-MPNN), was employed as the scoring function.
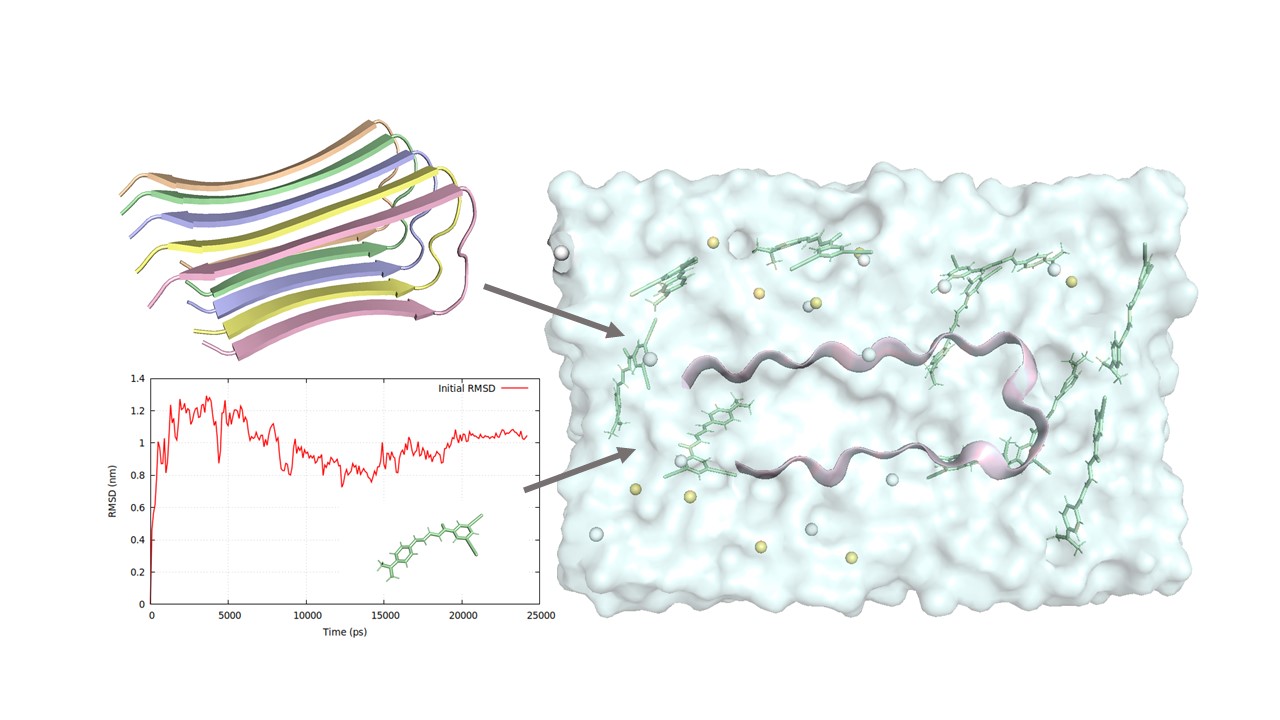
In the molecular modeling, the Aβ fibril model used in this study is based on the core structure of the Aβ42 fibril (PDB ID: 2BEG).6 All molecular dynamics simulations were performed with GROMACS 2024.3 software, utilizing the CHARMM36 force field. Both in-house python code and MDAnalysis tools were used for analyzing simulation trajectories.7 The steepest descent algorithm was used for energy minimization, followed by two equilibrations of 1 ns each under isothermal–isochoric (NVT) and isothermal–isobaric (NPT) conditions at room temperature. Production simulation runs were conducted in the isothermal–isobaric ensemble, applying the Nosé–Hoover thermostat and the Parrinello–Rahman barostat at 1 bar pressure for 1000 ns simulation time. Furthermore, steered molecular dynamics (SMD) with umbrella sampling was adopted for studying thermodynamic changes of the Aβ42 monomer.
Results:
The average Tanimoto index and Pearson correlation for structure similarity were used as evaluation metrics. Both evaluation metrics showed a positive correlation with high similarities between the compounds generated through the active learning iterative process and the original experimental molecules. When compared to other baseline models, i.e., Random generated sequence and BLOSUM generated sequence, the reinforcement learning method demonstrated superior performance in terms of Morgan fingerprints, RDKit descriptors, and sequence-based Quantitative structure-activity relationship (QSAR) similarities. Furthermore, similar stability of structural fluctuations was obtained in the interactions between the Aβ42 monomer and both the AI-generated anti-aggregation molecules and the top-performed experimental molecules. This was indicated by lower and more stable root-mean-square deviation (RMSD) and root-mean-square fluctuation (RMSF) values. The generated and the original experimental molecules also exhibited rapid and strong attractive interaction energies with the Aβ42 monomer. Additionally, the umbrella sampling combined with the weighted histogram analysis method (WHAM) revealed the transition from a folded U-shaped structure to an unfolded, nearly flat conformation. The result revealed a higher free energy profile for the Aβ42 monomer binding with AI-generated molecules compared to the top-performed experimental molecules.
Conclusions:
Our simulation results demonstrate that reinforcement learning successfully identified truly novel and diverse compounds with similar scaffolds to the experimental molecules and strong interaction affinities. This also highlights the importance of combining generative AI models with complementary physics-based molecular modeling methods. To evaluate and confirm this model, the generated Aβ42 anti-amyloidogenic molecules will be synthesized and tested in wet lab for comparison and validation. Furthermore, steered molecular dynamics simulations of anti-aggregation molecules and Aβ42 monomer provided insight into potential mechanisms for inhibiting Aβ formation and preventing aggregation. Overall, our findings indicate that combining generative AI with the molecular modeling approach can greatly accelerate small molecule discovery by efficiently narrowing the chemical search space.
Reference
- Loeffler, H. H.; Wan, S.; Klähn, M.; Bhati, A. P.; Coveney, P. V. Optimal Molecular Design: Generative Active Learning Combining REINVENT with Precise Binding Free Energy Ranking Simulations. Journal of Chemical Theory and Computation 2024, 20(18), 8308-8328.
- Duran, T.; Chaudhuri, B. Where Might Artificial Intelligence Be Going in Pharmaceutical Development? Molecular Pharmaceutics 2024, 21 (3), 993–995.
- Quiroz, J. P.; Zeng, A.; Young, M.; Gordon, P.; Aadya Jaipuria; Reed, K. J.; Landry, G. M.; Yang, S.; Asher, S.; Ruoyao, S.; Priefer, R. Homotaurine and Curcumin Analogues as Potential Anti-Amyloidogenic Agents. Chemistry 2023, 5 (1), 223–241.
- Aadya Jaipuria; Castillo, M.; Boksanski, J.; Landry, G.; Beak, J. H.; Young, M.; Priefer, D. T.; Kaïs Guessab; Ellis, C. N.; Priefer, R. Extended Chalcones: Synthesis, in Vitro Analysis, and in Vivo Testing against a Drosophila Melanogaster Alzheimer’s Disease Model. Chemistry 2024, 6 (6), 1477–1494.
- Loeffler, H. H.; He, J.; Alessandro Tibo; Jon Paul Janet; Alexey Voronov; Mervin, L. H.; Engkvist, O. Reinvent 4: Modern AI–Driven Generative Molecule Design. Journal of cheminformatics 2024, 16 (1), 20.
- Lemkul, J. A.; Bevan, D. R. Assessing the Stability of Alzheimer’s Amyloid Protofibrils Using Molecular Dynamics. The Journal of Physical Chemistry B 2010, 114 (4), 1652–1660.
- Liang, P.-Y.; Huang, X.; Duran, T.; Wiemer, A. J.; Bai, J. Exploring Latent Space for Generating Peptide Analogs Using Protein Language Models. 2024 IEEE International Conference on Bioinformatics and Biomedicine (BIBM) 2024, 842–847.