2025 AIChE Annual Meeting
(461b) Deciphering the Compensation Effect in Ammonia Decomposition Using First-Principles Kinetic Modeling
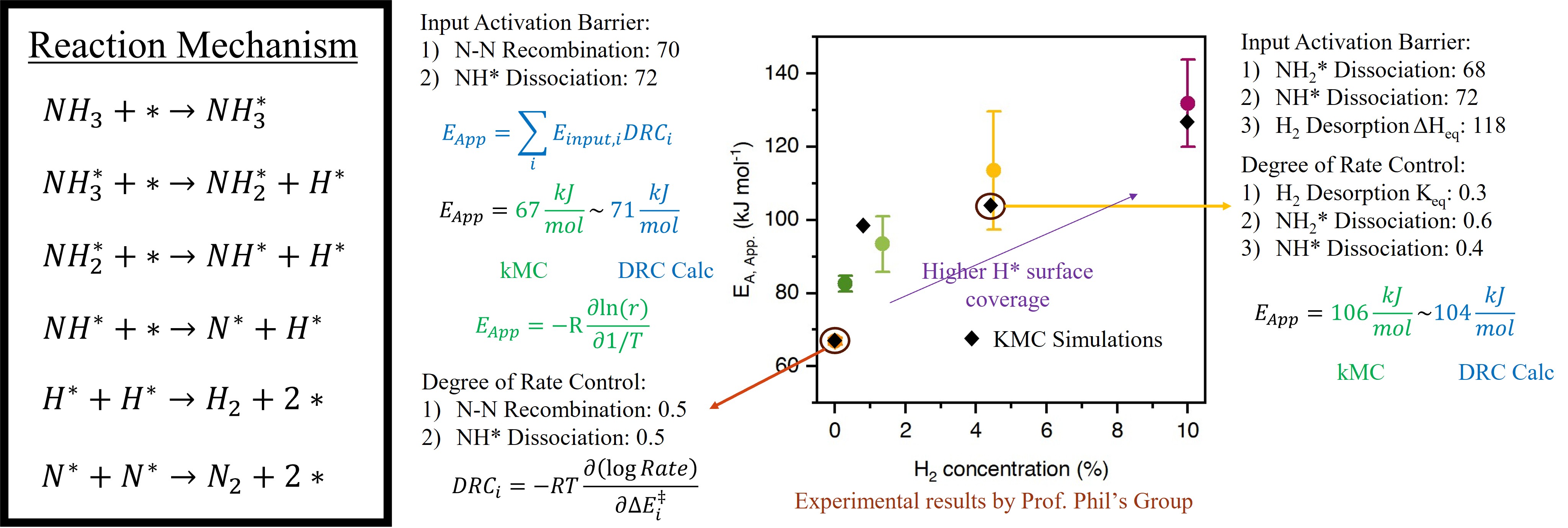
Herein, we employ a combined Density Functional Theory (DFT) and Kinetic Monte Carlo (kMC) simulations to provide mechanistic insights into the compensation effect. DFT calculations determine the kinetic parameters of the elementary steps and the binding energies of the adsorbates on Ru (0001) surface. The DFT calculated data was subsequently used to parameterize lateral interaction models and provide elementary kinetic database used in kMC simulations to simulate the reaction network as a function of reaction conditions.
kMC simulations have been performed at 0.01 bar NH3 pressure, temperature range of 573 – 773K and 0 – 0.1 bar H2 pressure on stepped Ru(0001) surface with explicit modeling of B5 sites. At 0 bar H2 pressure, the H* surface coverage is low, and the B5 sites are vacant to facilitate N-N recombination. Upon increasing H2 pressure, H* surface coverage increases and blocks the B5 sites, reducing the overall decomposition rate. Degree of Rate Control (DRC) analysis suggests that the DRC value of H2 desorption equilibrium constant increases from 0 at 0 bar H2 pressure to 0.3 at 0.05 bar H2 partial pressure. Since the heat of H2 desorption is positive and the preexponential factor is greater than one, the apparent barrier and the preexponential increases with increasing H2 pressure and therefore explains the observed compensation effect.
Overall, DFT and kMC simulations elucidate the compensation effect in NH3 decomposition, showing that increased H2 partial pressure leads to higher H* coverage, poisoning step-edge sites, reducing the decomposition rate, and driving the observed compensation behavior.