2025 AIChE Annual Meeting
(199f) Contrasting Bi2O3 and V2O5 Catalysts for the Initial C–H Activation of C2H6 and the Selective Hydrogen Combustion Reactions
Authors
Rajeev Kumar - Presenter, Purdue University
Jacob Rothman, University of Florida
Randall Meyer, Exxonmobil
David Hibbitts, University of Florida
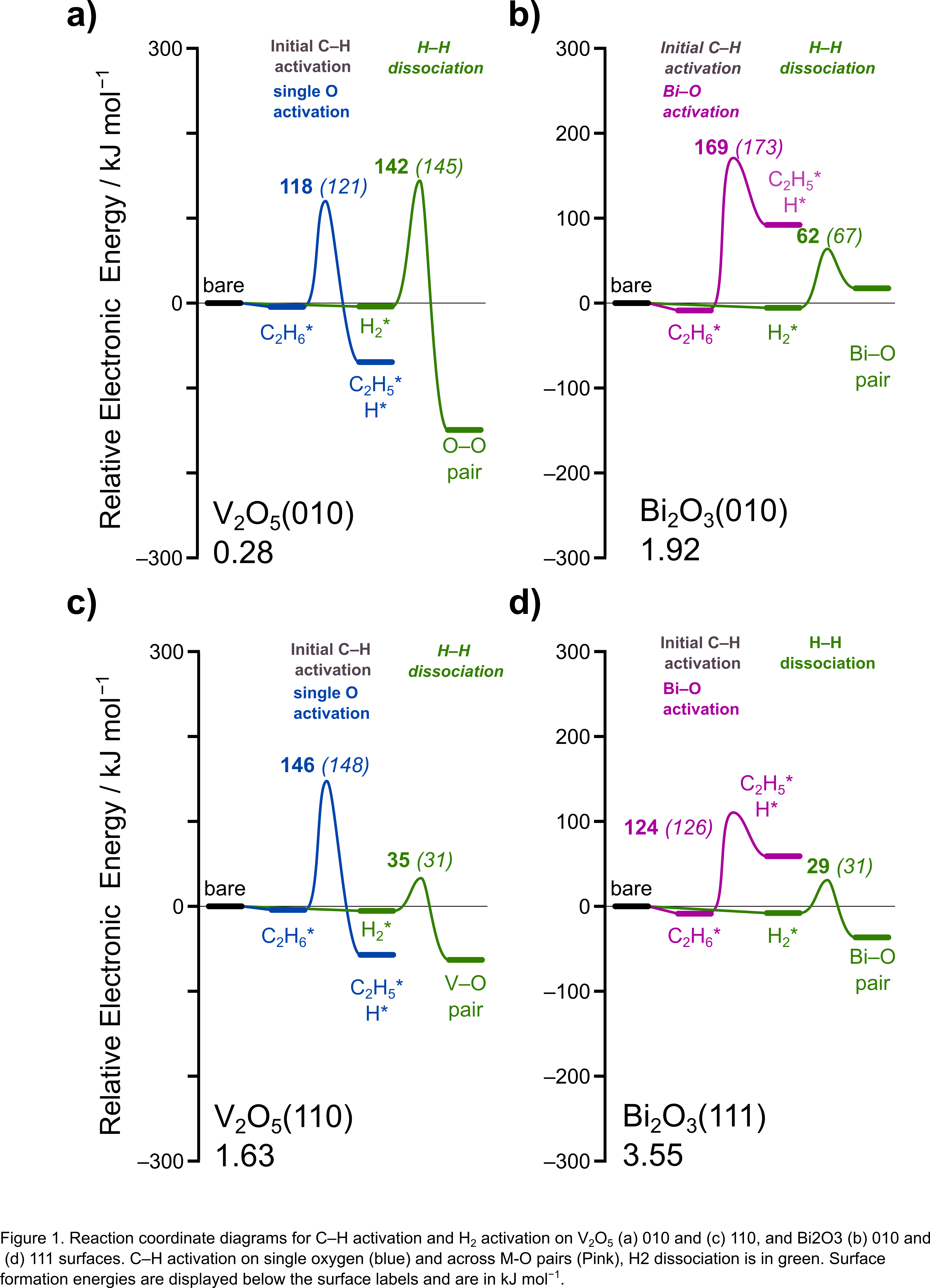
Alkane dehydrogenation via direct and oxidative pathways has emerged as an alternative method to steam cracking for alkene production. The direct dehydrogenation pathway is highly endothermic and prone to catalyst deactivation through coke formation, while the oxidative pathway is exothermic but suffers from poor selectivity and high COx formation. Overcoming these challenges, selective hydrogen combustion (SHC) combines a direct dehydrogenation catalyst with one that can selectively combust hydrogen, achieving the same overall thermodynamics as oxidative dehydrogenation. This approach helps improve the yield and selectivity of alkene formation while reducing the energy costs associated with the endothermic dehydrogenation step. Here, we use density functional theory (DFT) calculations to compare the properties of Bi2O3, a known SHC catalyst, to V2O5, a known alkane combustion catalyst. These reactions can be carried out in a steady-state mode or in chemical looping mode, where dehydrogenation and SHC occur in tandem or a sequential bed, forming the desired alkene and H2O while reducing the SHC catalyst. The SHC catalyst is reoxidized to complete the loop in a subsequent step using molecular O2. We compare H2 dissociation and alkane activation barriers on multiple surfaces of Bi2O3 and V2O5. Additionally, we calculate common reducibility descriptors, such as oxygen vacancy formation energies (VFE), hydrogen addition energies (HAE), and methyl-addition energies (MAE). We show that coordinatively unsaturated (cus) metal atoms, common on Bi2O3 but rare on V2O5, can bind hydrides and promote H2 dissociation across unbound M,O pairs. Higher selectivity can be achieved if the SHC catalyst's O atoms exhibit relatively weak H-abstraction, making them less effective at activating hydrocarbons. Cus metal atoms that bind hydrides and oxygen atoms with relatively weak HAE energies—contribute to the high rates of H2 combustion relative to alkane combustion on stoichiometric oxide surfaces.