2025 AIChE Annual Meeting
(253a) Comparison of Surface-Mediated and Proton-Coupled Electron Transfer Pathways for Formation of C-H Bonds in Electrocatalytic Reduction Reactions
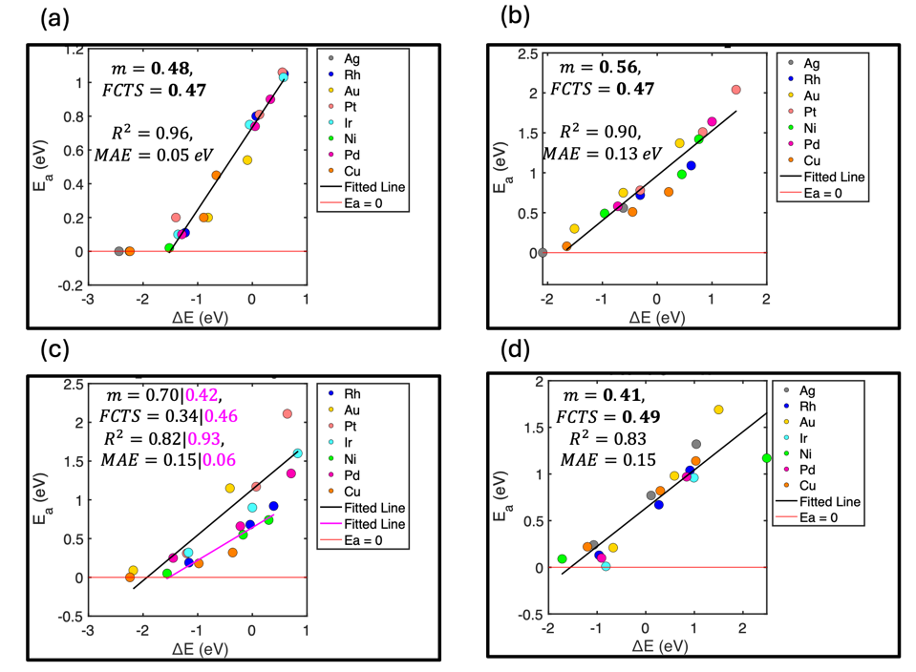
To provide insights into electrochemical hydrogenation trends across transition metal surfaces, and to compare the energetics of both surface-mediated and proton-coupled electron transfer reactions under these conditions, we employ the rigorous cell extrapolation method to evaluate the constant potential activation barriers and reaction energies for both protonation reactions (C* to CH*, CH* to CH2*, CH2* to CH3*, and CH3* to CH4) and the Volmer reaction across a series of eight transition metal (111) surfaces: Ag, Au, Ir, Pt, Pd, Ni, and Rh. We developed a series of electrochemical BEP relationships from these results, and we determined that the fractional coordinate of the transition state (FCTS, xTS) correlates well with the slopes of the BEP relationship for these electrocatalytic chemistries. Thus, knowing the xTS of a protonation reaction on a single metal can serve as a predictor for the BEP slope, enabling the estimation of activation energies on other metals and at varying potentials. Furthermore, we compare the BEP relationships for surface hydrogenation reactions with those of electrochemical protonation reactions. These findings contribute to a better understanding of the relative roles of surface and PCET-catalyzed reaction steps in electrochemical environments and may ultimately facilitate the transfer of knowledge from elementary reactions to industrial electrocatalytic systems.