2025 AIChE Annual Meeting
(390ay) Chemically-Informed Machine Learning Approach for Prediction of Reactivity Ratios in Radical Heteropolymerization
Authors
Chemically-Informed Machine Learning for Predicting Reactivity Ratios in Radical Heteropolymerization
Understanding and predicting monomer reactivity ratios is crucial for controlling heteropolymer chain configurations and their resultant properties. Traditional experimental determination of these ratios is time-consuming and resource-intensive, while existing computational methods often struggle with accuracy or scalability. In this work, we present a novel hybrid approach that combines unsupervised learning with deep neural networks to predict reactivity ratios in radical copolymerization.
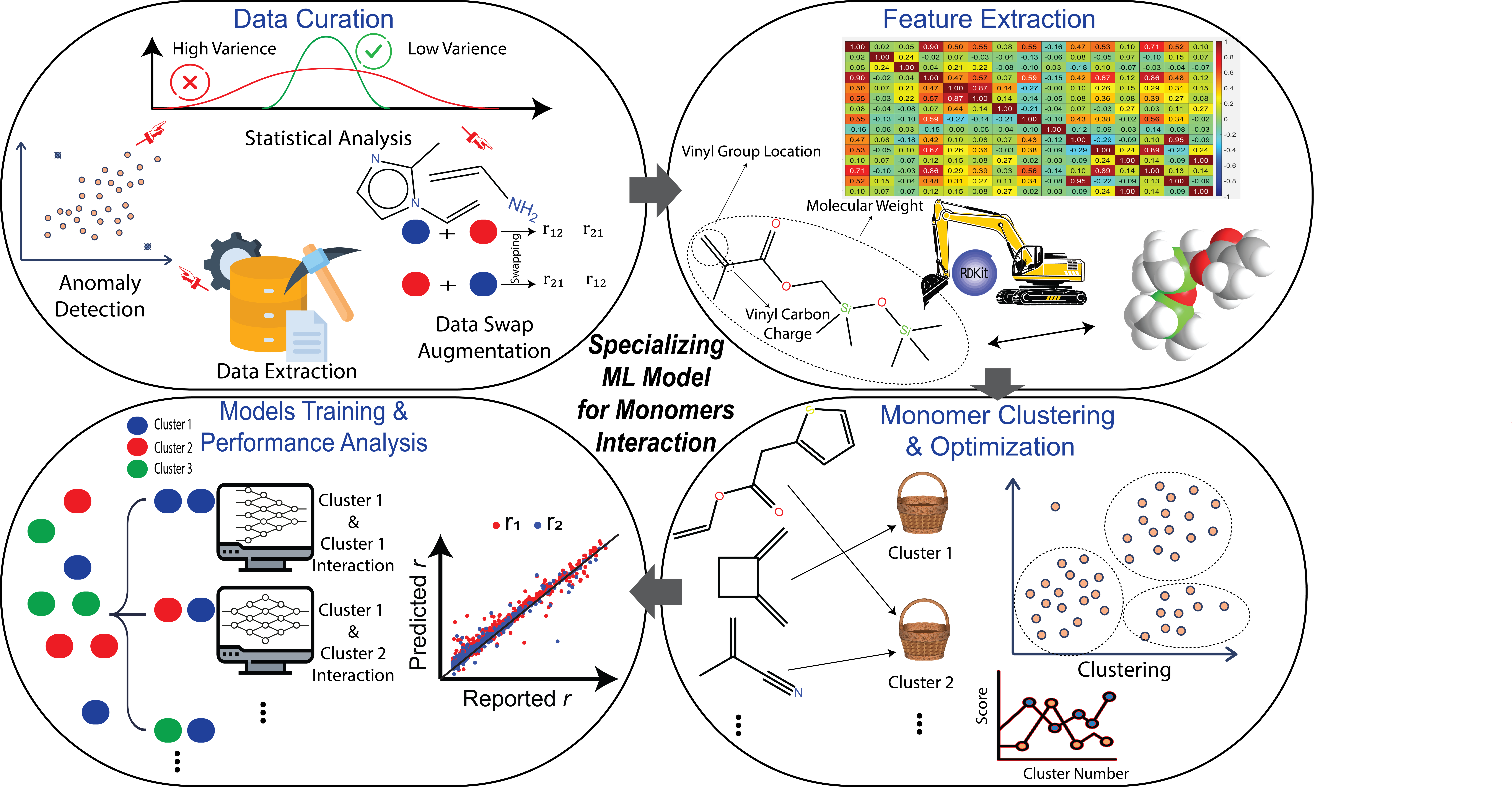
Our approach begins with comprehensive feature extraction from monomer structures. We identified eight key physicochemical properties that influence radical polymerization behavior: molecular weight, vinyl position (cyclic or linear), vinyl carbons charge, sp² hybridization, total polar surface area, molecular LogP, number of hydrogen donors, and stereochemistry. These features were selected through rigorous correlation analysis and chemical reasoning to ensure they captured distinct aspects of monomer reactivity while minimizing redundancy.
Statistical analysis of our dataset revealed significant differences in reactivity based on structural features. For instance, monomers with unringed vinyl groups showed substantially higher reactivity (mean = 1.35) compared to those with ringed vinyl groups (mean = 0.67). This observation aligns with mechanistic understanding, as vinyl groups embedded in ring structures face greater steric constraints and require higher energy for ring-breaking during propagation.
By applying spectral clustering to these molecular features, we identified three distinct monomer groups with characteristic reactivity patterns. Cluster 1 comprised monomers with intermediate characteristics and higher reactivity values. Cluster 2 included monomers characterized by vinyl groups within rings, high vinyl carbon charge, elevated total polar surface area, and fewer hydrogen donors. Cluster 3 was distinguished by monomers with high sp² hybridization counts, larger molecular weights, and increased lipophilicity.
The clustering process involved comprehensive evaluation of five different algorithms (k-means, Spectral Clustering, Agglomerative Clustering, Gaussian Mixture Model, and BIRCH) across multiple evaluation metrics (silhouette score, Calinski-Harabasz index, and Davies-Bouldin index). Spectral clustering with three clusters emerged as the optimal choice, demonstrating consistently high silhouette scores (0.95-1.0) and uniform performance across all evaluation criteria.
Analysis of reactivity ratios within and between clusters revealed distinct patterns in monomer interactions. Monomers in Cluster 1, when interacting with themselves, demonstrated the highest average reactivity ratio (1.643) but also exhibited significant variability (variance = 10.618). This suggests a high tendency to create block-type copolymers. In contrast, Cluster 2 showed notably low reactivity (mean = 0.077) and minimal variance (0.008), indicating a strong preference for alternating sequences. Cluster 3 maintained moderate reactivity (mean = 0.983) conducive to random copolymerization.
For our predictive models, we implemented deep neural networks optimized through Bayesian hyperparameter optimization. Morgan fingerprints with a radius of 3 and 2048-bit vectors were used to convert monomer structures to machine-readable format. The optimized architecture consisted of three hidden layers (654, 305, and 51 neurons) with ReLU activation functions, dropout layers (rate of 0.29), and L2 regularization (λ=0.0028).
We evaluated the impact of cluster-specific training by comparing two approaches: a general model trained on the complete dataset and specialized models trained exclusively on interactions between specific cluster pairs. The results demonstrated substantial improvements in prediction accuracy with the cluster-specific approach. For Cluster 1-Cluster 1 interactions, the R² values increased to 0.743 for r₁ and 0.644 for r₂, compared to 0.433 and 0.319 respectively for the non-clustered model. Even more remarkable improvements were observed for Cluster 1-Cluster 3 interactions, with R² values of 0.637 and 0.902 for r₁ and r₂.
This significant enhancement in prediction accuracy can be attributed to several factors. First, the clustering approach effectively reduces the chemical space complexity by grouping monomers with similar reactivity patterns. This specialization allows the model to focus on more consistent structure-reactivity relationships within each cluster interaction type. Second, when the dataset is specialized for specific cluster interactions, the distribution of reactivity ratios becomes more normalized, leading to better learning outcomes.
The higher R² values observed for Cluster 1-Cluster 3 interactions compared to Cluster 1-Cluster 1 interactions suggest that inter-cluster interactions may exhibit more distinct and learnable patterns than intra-cluster interactions. This observation aligns with chemical intuition that monomers from different clusters would have more differentiated reactivity patterns, making their interactions more predictable once properly categorized.
Our chemically-informed machine learning framework demonstrates that pre-clustering of chemical data before applying predictive models can be a powerful strategy in polymer informatics, particularly when dealing with complex structure-property relationships. This approach not only enhances prediction accuracy but also provides valuable insights into the underlying chemical mechanisms governing heteropolymerization.
The ability to accurately predict reactivity ratios enables more efficient polymer synthesis planning and facilitates the discovery of new heteropolymers with tailored properties. By understanding which monomer combinations are likely to produce specific chain configurations (alternating, random, or block-type), researchers can strategically select monomers to achieve desired material properties, significantly reducing the experimental effort required for materials development.
This work represents a significant advancement in computational approaches to polymer design and synthesis optimization. The integration of chemical knowledge with machine learning techniques addresses the limitations of both traditional experimental methods and pure computational approaches, offering a more efficient pathway for the development of novel polymer materials with tailored properties.