2025 AIChE Annual Meeting
(675g) Bulk Descriptors for Predicting O and OH Adsorption Energies on Metal Oxides across Varied Coordination Environments
Authors
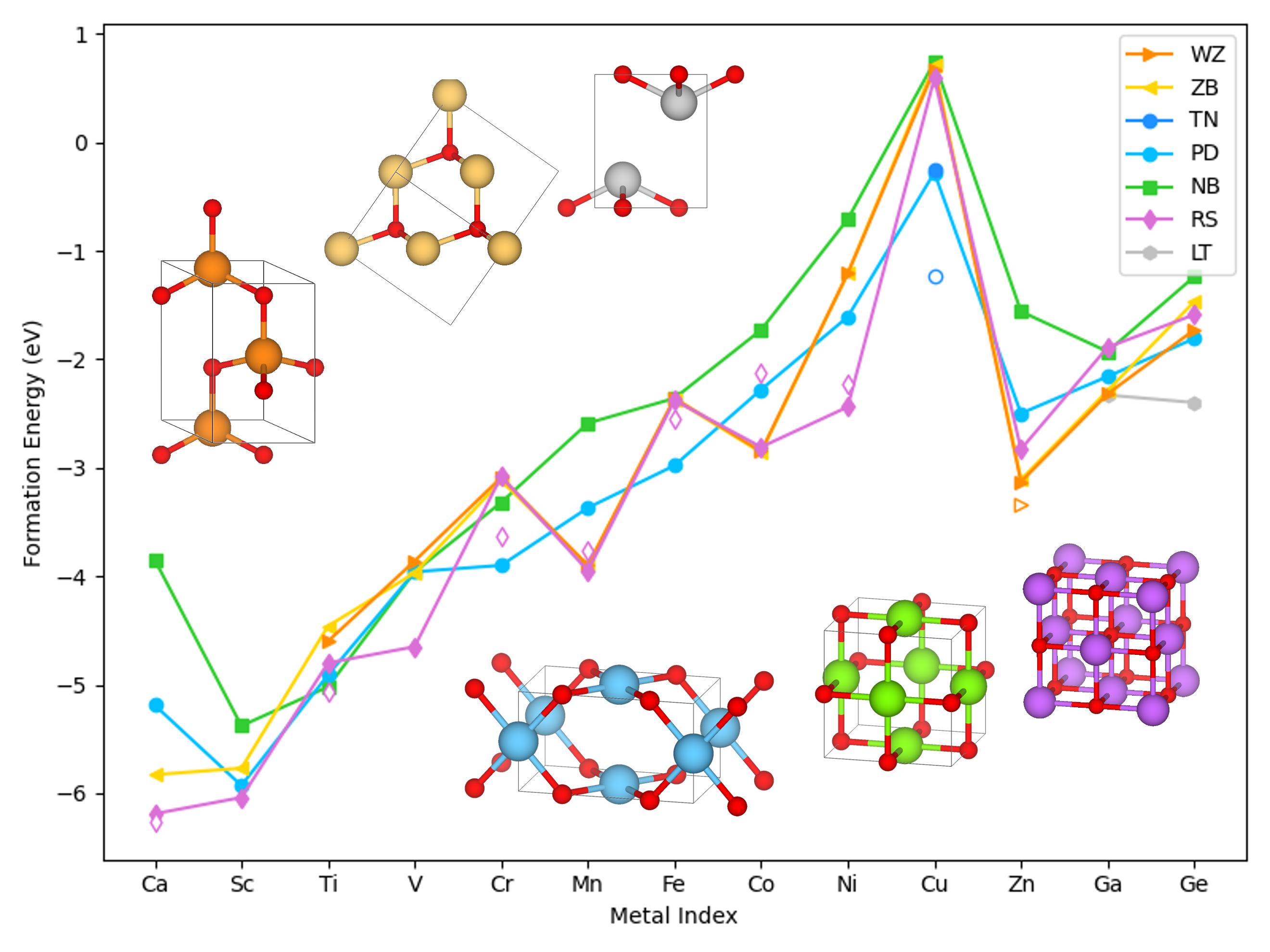
Before applying machine learning, we analyzed a dataset of over 300 DFT-optimized binary metal oxides to extract physical trends. Bulk formation energies revealed characteristic d-electron effects: a V-shaped profile with a node at d¹ metals across the transition series, and an M-shaped curve centered on Mn within the 3d row—mirroring crystal field splitting behavior. The calculated formation energies and the most favorable coordination types also showed strong agreement with experimental reports, reinforcing the reliability of the dataset.
Using a combination of DFT-based descriptors (e.g., ICOHP, Bader charge) and elemental features from the Mendeleev library, we trained multiple regression models. Gaussian Process Regression yielded the best accuracy (MAE = 0.2591 eV, MSE = 0.1133 eV²), outperforming gradient boosting, random forest, and linear regression. Feature importance analysis highlighted the need to account for both covalent and ionic bonding characteristics. Classification models predicting coordination environment achieved 76% accuracy when grouped by geometry type.
This work also includes adsorption energy predictions for *O and *OH species on representative surfaces, enabling a direct connection between bulk-derived descriptors and surface reactivity. By integrating electronic structure insight with machine learning, we provide a data-efficient framework for exploring catalytic trends across structurally diverse oxide materials—without relying on explicit surface calculations.