2025 AIChE Annual Meeting
Bridging Visual Chemistry and Language Intelligence: Extracting Scattered Information from Chemical Reaction Mechanism Images into Structured Data
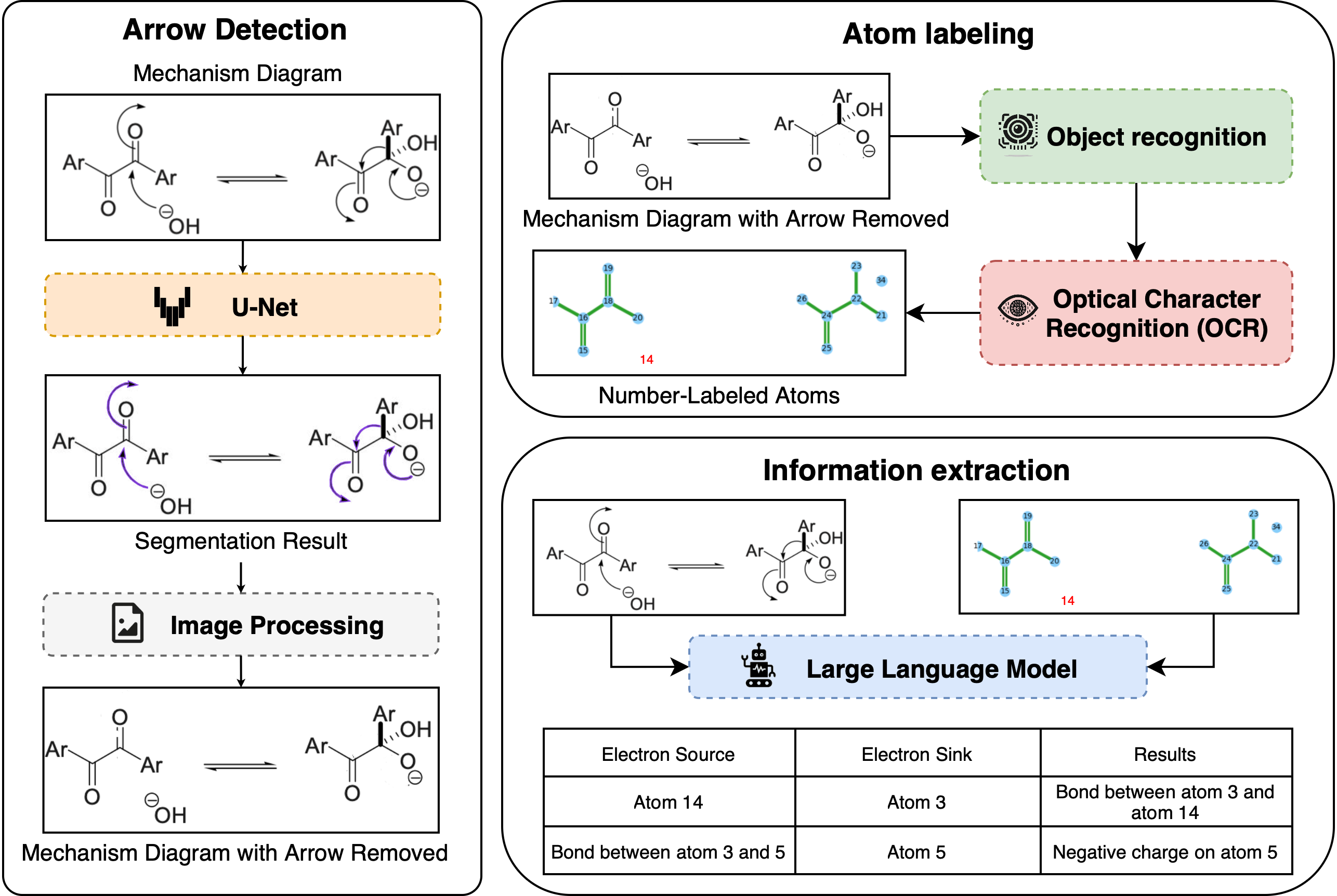
This project introduces a complete system in order to address such problems. This system processes chemical reaction images through a series of steps: segmenting and removing curved arrows, recognizing molecular structures, identifying reaction components, labeling atoms for tracking, and interpreting reactions with large language models (LLMs). Each part of the pipeline was designed and tested to ensure it supports accurate extraction of chemical information from complex diagrams.
The initial stage utilizes a U-Net-based segmentation model, specifically trained to detect and remove curved arrows from chemical images. This preprocessing step significantly enhances the clarity of molecular diagrams, proven by a substantial increase in recognition accuracy. For instance, testing on a dataset of reaction mechanism images revealed improvements in exact SMILES matching from 12.1% to 64.5% using the MolScribe recognition model [1]. Similar performance gains were observed with DECIMER, another prominent chemical recognition tool [2, 3], further validating the effectiveness of our preprocessing strategy. Subsequent stages involve extracting and labeling individual reaction components and assigning numerical identifiers to atoms for precise referencing. These structured annotations enable effective interpretation by large language models (LLMs), facilitating accurate mechanistic analysis. In comprehensive benchmarking using a synthetic dataset containing 17,497 annotated reaction images, structured atomic referencing increased descriptive task accuracy from 7.3% to 79.5% and interpretive accuracy from 6.2% to 68.4%. The final phase involves using LLMs for mechanism-guided reaction prediction, achieving notable performance for single-step reaction diagrams in exact SMILES accuracy of 19.3% and molecular graph reproducibility of 17.2%. This demonstrates the pipeline's potential for accurate, automated extraction and interpretation of chemical reaction mechanisms. Alongside the development of the main pipeline, the work also delivered three pioneering dataset regarding molecular identities, reaction information, and electron movements.
Our approach not only addresses a longstanding challenge in chemical informatics but also significantly enhances the accessibility of chemical data for computational applications, all critical to modern chemical engineering and research [4]. By transforming visually complex chemical diagrams into structured datasets, this work provides a significant advancement in computational process simulation and chemical reaction optimization.
References:
[1] Y. Qian, J. Guo, Z. Tu, Z. Li, C. W. Coley, and R. Barzilay, ‘MolScribe: Robust Molecular Structure Recognition with Image-to-Graph Generation’, J. Chem. Inf. Model., vol. 63, no. 7, pp. 1925–1934, Apr. 2023, doi: 10.1021/acs.jcim.2c01480.
[2] K. Rajan, A. Zielesny, and C. Steinbeck, ‘DECIMER 1.0: deep learning for chemical image recognition using transformers’, Journal of Cheminformatics, vol. 13, no. 1, p. 61, Aug. 2021, doi: 10.1186/s13321-021-00538-8.
[3] K. Rajan, A. Zielesny, and C. Steinbeck, ‘DECIMER: towards deep learning for chemical image recognition’, Journal of Cheminformatics, vol. 12, no. 1, p. 65, Oct. 2020, doi: 10.1186/s13321-020-00469-w.
[4] K. M. Jablonka, P. Schwaller, A. Ortega-Guerrero, and B. Smit, ‘Leveraging large language models for predictive chemistry’, Nat Mach Intell, vol. 6, no. 2, pp. 161–169, Feb. 2024, doi: 10.1038/s42256-023-00788-1.