2025 AIChE Annual Meeting
(203k) Bridging Timescales in Protein Simulations Using Parallelized Gaussian Accelerated Molecular Dynamics (ParGaMD)
Authors
Anugraha Thyagathur, University of California, Davis
Maxen Hamelynck, University of California, Davis
Roland Faller, Texas Tech University
Surl-Hee Ahn, University of California, Davis
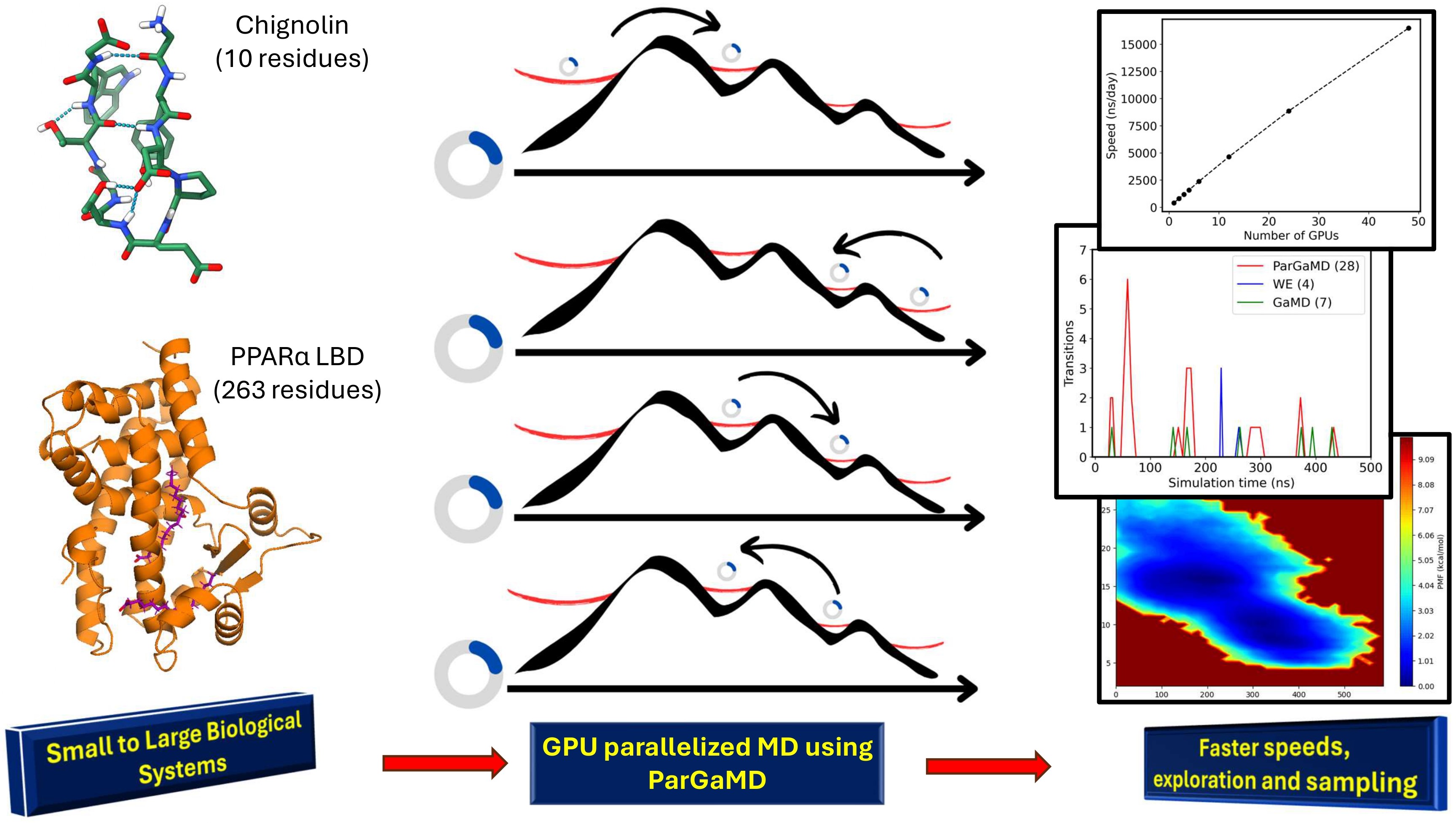
Molecular dynamics (MD) simulations have been used as a computational microscope to investigate and understand phenomena related to protein dynamics. However, since MD simulations are run using 10−15 s time steps due to being limited by the fastest motions in the system (e.g., the vibration of bonds) and protein dynamics ranges from 10−6 s to longer, there is a major timescale barrier between MD simulations and protein dynamics. Though “enhanced sampling methods,” a class of computational tools, can help accelerate the exploration of the protein conformational space and dynamics compared to conventional MD simulations, obtaining accurate thermodynamic and kinetic properties of large systems (>200,000 atoms) from MD simulations in a computationally tractable period is still challenging. To tackle this issue, we developed a novel enhanced sampling method called “parallelized Gaussian accelerated molecular dynamics” (ParGaMD), which runs many short Gaussian accelerated molecular dynamics (GaMD) simulations over multiple GPUs in parallel by using the weighted ensemble method (WE) framework. Although GaMD has demonstrated accelerated sampling for many systems by adding a boost potential to the system, GaMD takes weeks to run for large systems since the Amber MD GPU engine pmemd.cuda, in which GaMD is mainly implemented, does not parallelize well over multiple GPUs. By using the WE framework, we were able to overcome this bottleneck and sample more efficiently along the chosen collective variables, which enables ParGaMD to be more powerful than GaMD itself. ParGaMD can significantly accelerate the sampling of different conformational states and dynamics of any protein, biomolecule, and material system, which will benefit the wider scientific community. We will demonstrate that ParGaMD is able to achieve close to linear scaling on multiple GPUs over two popular MD engines (AMBER and OpenMM) and significantly reduce the needed wall time for MD simulations of various biomolecular systems.