2025 AIChE Annual Meeting
(168e) AI-Powered Classification of iPSC-Derived ? Cells Using Microscopic Images
Authors
With the advancement of regenerative medicine to clinics and the applications of hPSC-derived tissues and organs in regenerative therapy and in-vitro disease models, there is a need to generate functional organoids of high quality and with high consistency. While significant progress has been made in the functional maturation of most of the hPSC-derived organoids, it has been challenging to remove heterogeneity from the process of differentiation. Thus, the organoid generation pipeline must undergo stringent quality testing, which slows the process because most quality assessment methods are invasive, damaging, and manual. In this project we have developed a Deep Learning model to perform single cell quality assessment solely based on brightfield images. Hence, this process can be conveniently integrated with the differentiation pipeline while preserving the efficiency and quality of differentiation. Convolutional neural networks (CNNs) can allow cell image classification through the automation of cellular phenotype detection from microscopic images. However, the training of accurate classification models requires a large dataset, which can be challenging. Synthetic data, which can be possible through generative AI (GenAI) processes, has been proposed as a solution to enrich training datasets to raise classification accuracy. Here, we present an innovative and non-invasive technology that helps us not only to classify desirable hiPSC-derived insulin producing β cells, but also to generate microscopic images (brightfield). We hypothesize that brightfield images of β cells derived from hiPSCs contain adequate information, which can be extracted using convolutional neural network (CNN), to establish the unique morphometric signatures associated with differentiation. We developed a very accurate CNN model based on brightfield images that achieved a remarkable accuracy rate of around 92% for insulin-producing cell classification. In addition, we will use GenAI methodologies to develop a model capable of generating brightfield images of β cells. These developments will highlight the capacity of AI-driven cell image classification and data generation as a link between biological knowledge and computational capability.
Methods:
Cell Staining:
Two stains targeting insulin-producing cell markers defined iPSC-differentiated cells. Primary antibody rabbit anti-NKX6.1, a transcription factor essential for β cell development, and secondary antibody anti-rabbit Alexa Fluor 488, which fluoresces green. Second stain targeted C-peptide, a result of insulin synthesis, verifying insulin production using mouse anti-C-peptide as a primary antibody. The secondary antibody was anti-mouse Alexa Fluor 647.
Image Acquisition
To analyze the differentiated cells, we utilized an imaging flow cytometer, specifically the ImageStream, to capture both brightfield and fluorescent images. We used the ImageStream Mark II, INSPIRE 744 using 60X magnification for the quantification of protein expression. The gate was positioned downstream of cells that were positive just for the secondary antibody to exclude any potential false positive results.
Hardware Specifications
For building the AI models, the hardware was equipped with a processor 16-Core 3.90 GHz (AMD RYZEN Threadripper Pro 3955WX), memory 128 GB (4 x 32 GB), Graphics Card (1 x NVIDIA RTX 3090 Ti 24GB). For high-performance GPU-accelerated software environments, the NVIDIA CUDA Toolkit 11.5 (NVIDIA Corp., Santa Clara, CA, USA) was built on Ubuntu 22.04.4 LTS (Canonical Ltd., London, UK) with kernel version 6.5.0-25-generic.
Evaluation of CNN Performance
The evaluation between the output prediction and the target was conducted using accuracy, recall, precision, F1-measure, Mathews Correlation Coefficient (MCC), and the area under the receiver operating characteristic curve (AUCROC). Grad-CAM was used to visualize important regions of single cell images.
Results:
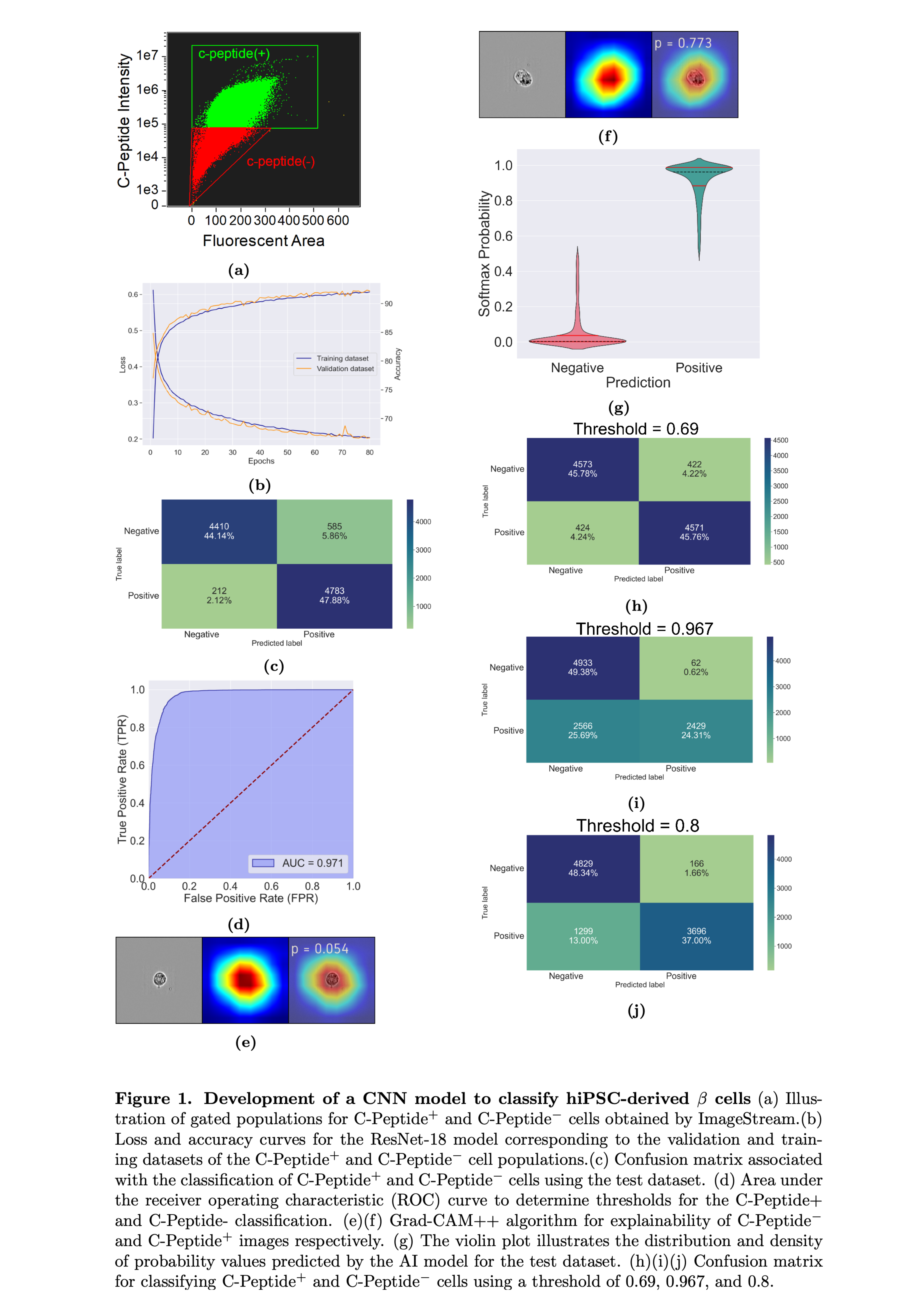
The cells were stained for c-peptide and NKX-6.1 after fixation. We selected brightfield images from fluorescent images to identify positive group cells using these markers. The cell populations were manually selected using the expression of the markers and then grouped into C-Peptide+ and C-Peptide- classes (Figure 1a). The model was trained using a total of 99,886 brightfield images for the classification of positive and negative C-Peptide cells. For training the neural network, 39,954 images were used per class, while 4,994 images per class were used for model validation during training. Additionally, another 4,995 images per class were used to evaluate the trained model. The images were enlarged to dimensions of 224 by 224 by replicating the last row and column of pixels in each image to fill the extra border. The construction of the CNN model required a batch size of 32, a learning rate of 0.00001, and a momentum of 0.9. The trained CNN model exhibited loss levels nearing 0.2 and no discrepancy between the loss values in the training and validation data (Figure 1b). After undergoing training, the CNN model exhibited strong performance when tested with the dataset. The confusion matrix reveals that the model accurately classified 4,783 (47.88%) images as positive, 4,410 (44.14%) images as negative, misclassifying 212 (2.12%) images as negative, and producing 585 (5.86%) false positive images. Hence, the model attained an accuracy of 0.92, an F1 score of 0.92, an MCC of 0.84, precision of 0.89, recall of 0.96, and ROC-AUC of 0.971. (Figure 1c, d). The Grad-CAM analysis revealed that the model effectively detected both positive and negative cells by precisely recognizing important characteristics and patterns present in the cell images (Figure 1e, f). Following the development of the C-Peptide classification model, we assessed its performance at various threshold levels. These thresholds were selected by a descriptive statistical analysis, based on the probability score for the positive class of the test dataset. Based on the median, mean, and 75th percentile, the threshold values are 0.69, 0.52, and 0.967 respectively (Figure 1g). At the threshold of 0.69, the confusion matrix indicates that the model correctly identified 4,571 (45.76%) images as positive and 4,573 (45.78%) images as negative. However, it misclassified 424 (4.24%) images as negative and produced 422 (4.22%) false positive images (Figure 1h). The model achieved an accuracy of 0.92, an F1 score of 0.92, an MCC of 0.83, a precision of 0.92, and a recall of 0.92. When the threshold is set at 0.967, the confusion matrix shows that the model accurately classified 2,429 (24.31%) images as positive and 4,933 (49.38%) images as negative. Nevertheless, it incorrectly categorized 2566 (25.69%) images as negative and generated 62 (0.62%) false positive images (Figure 1i). The model attained an accuracy of 0.74, an F1 score of 0.65, an MCC of 0.55, a precision of 0.98, and a recall of 0.49. With a threshold of 0.8, the confusion matrix suggests that the model correctly identified 3,696 (37.0%) images as positive and 4,829 (48.34%) images as negative. However, it erroneously classified 1,299 (13.0%) images as negative and produced 166 (1.66%) false positive images (Figure 1j). The model achieved an accuracy of 0.85, an F1 score of 0.84, an MCC of 0.73, a precision of 0.96, and a recall of 0.74.
Implications:
We demonstrated that Convolutional Neural Network models can be effectively trained to categorize β cells produced from induced pluripotent stem cells using brightfield images. These images provide valuable morphological information by capturing aspects like shape and textures. The performance of this model was assessed using a separate dataset, resulting in a classification accuracy of 92% for distinguishing between positive and negative classes based on the C-Peptide and NKX-6.1 markers. Furthermore, we employed the MCC (Matthews Correlation Coefficient) and F1 score to assess the effectiveness of the model. The F1 score of 0.92 indicates that the model is highly effective in accurately classifying positive cells compared to other cases in the dataset. Despite F1 score is a remarkable indicator for accuracy, we have opted to utilize a more robust statistical measure for binary classification. The Grad-CAM algorithm has once again confirmed that the model selects the most relevant pixels to make its decision by utilizing those that are located within the cell. We evaluated the model at different threshold levels to analyze its accuracy and false positive classification. When the threshold was adjusted to the median value of 0.69, the model's accuracy, F1 score, and MCC values remained comparable to those achieved at a threshold of 0.5. Nevertheless, there was a marginal rise in the precision value of around 0.03, suggesting a modest decrease in the occurrence of false positives of around 1.6%. Similarly, adjusting the threshold to the 75th percentile value of 0.967 resulted in a considerable fall in the model's accuracy, F1 score, and MCC values compared to the values reached at a threshold of 0.5. However, there was a notable increase in the precision metric of around 0.09, indicating a substantial reduction in the occurrence of false positives by approximately 5.24%. Finally, we selected a threshold value of 0.8, which led to a moderate improvement in the model's accuracy but significantly reduced the occurrence of false positive outcomes by approximately 4.2%. With brightfield microscopy images and machine learning algorithms, it is possible to detect distinct morphological characteristics to classify specialized cell types (differentiation). In this project, we demonstrate that an AI model can identify distinct features associated with differentiation that are not detectable by human vision, impacting significantly the field of stem cells by enabling label-free identification and phenotype detection. This technique could highlight the potential for scalable cell therapies, improved disease modeling, or advancements in imaging techniques for protein quantification.