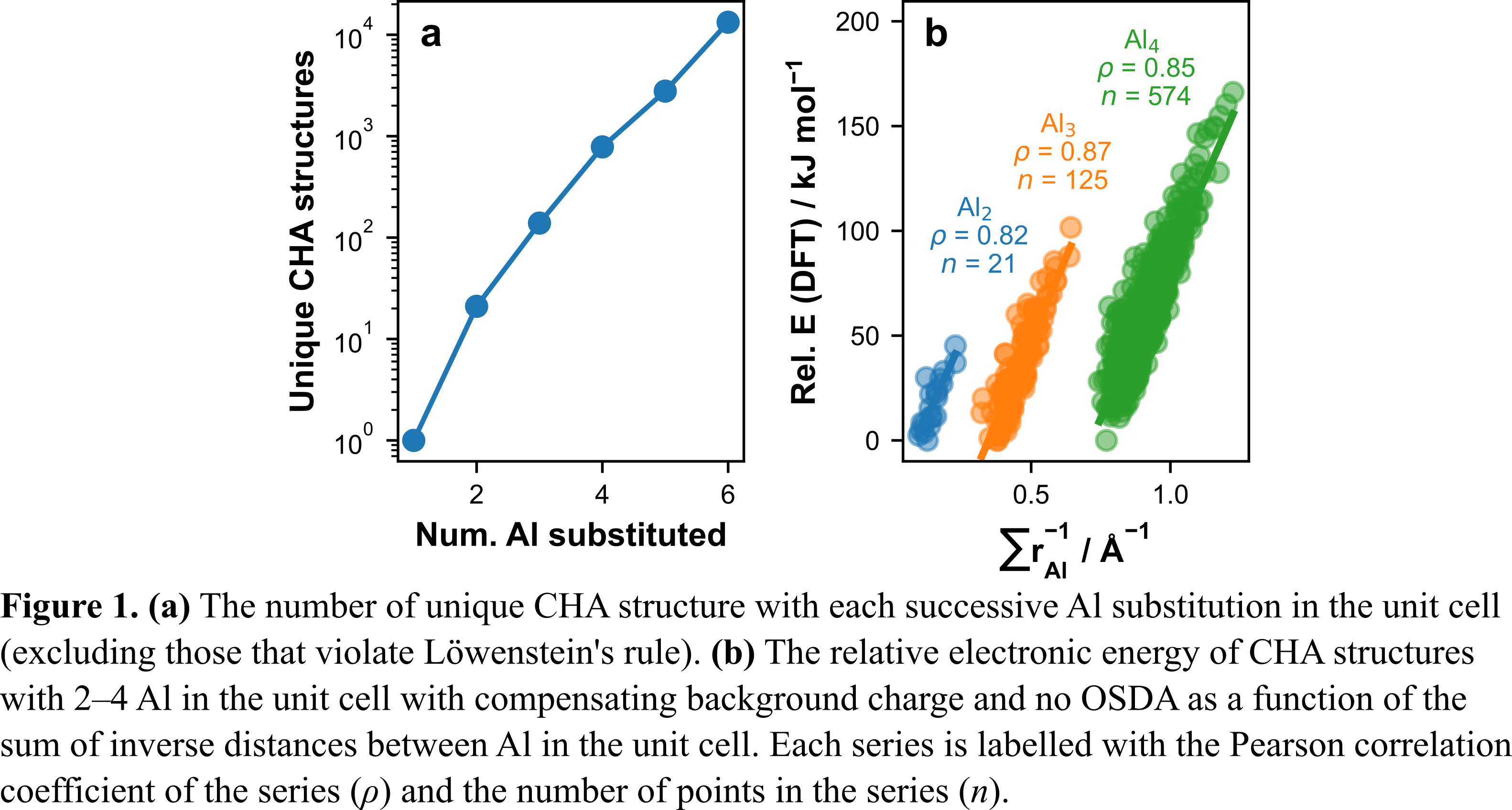
Zeolites are porous aluminosilicate materials that are commonly used as catalysts. Substituting Al
3+ for Si
4+ introduces a net anionic charge. This anionic charge can be balanced by H
+ to produce a Brønsted acid site, among other cations. Changing the distribution of framework Al in zeolites can change turnover rates (per H
+) for some reactions [1, 2]. Despite the role of Al distributions in catalysis, the factors that govern it are not fully understood. Recent findings showed that the relative stability of the crystallographic position of the Al in a framework and a sum of coulombic interactions between cationic organic structure-directing agents (OSDAs) and Al influenced Al placement [3]. Here, we enumerate all unique Al arrangements in CHA zeolites for Si/Al=5–35 (1–6 Al substitutions in a 36 T-atom unit cell). There is only one symmetrically unique Al position in CHA, but the number of unique structures increases with each Al substitution (Fig. 1a). Therefore, Al positioning drastically alters the number of symmetrically equivalent structures even in high-symmetry frameworks and brute-force introduction of Al without removing duplicate structures can make rigorous computational studies combinatorically infeasible. We compute the energies of these arrangements with and without cationic OSDAs balancing framework anions using density functional theory (DFT). Energies of structures without OSDAs depend linearly on the sum of inverse Al-Al distances in the unit cell (Fig. 1b), indicating that predominantly coulombic interactions govern the relative stability of Al arrangements. Finally, we use these data and similar calculations with cationic OSDAs present to train a graph neural network model to evaluate the stabilities of Al arrangements with cations present without costly DFT calculations.
References
[1] Chem. Mater. 2016, 28, 2236–2247.
[2] ACS Catal. 2017, 7, 6663–6674.
[3] Chem. Mater. 2020, 32, 9277–9298.