2024 AIChE Annual Meeting
(514e) Demystify Chemical Reaction Mechanisms Using Machine-Learning Enabled Thermochemistry Estimator (METE)
Authors
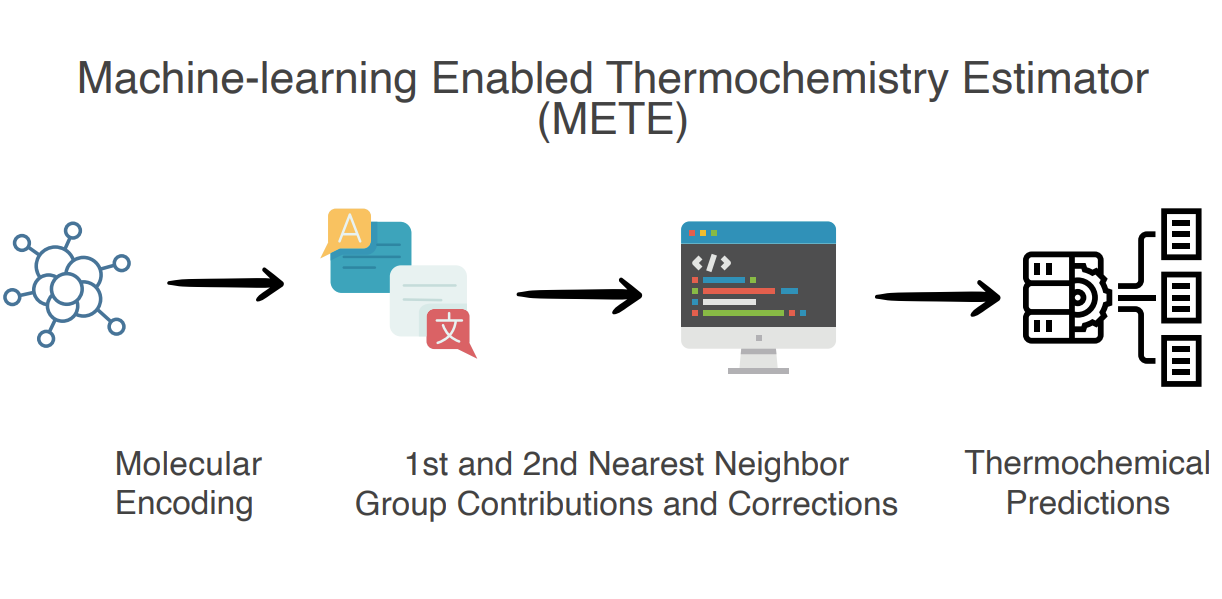
We establish an innovative framework to overcome this limitation, allowing efficient and accurate predictions of thermodynamical properties, such as enthalpies and Gibbs free energies. We utilize the framework leveraging techniques including text processing, descriptor featurization, and machine learning to replace costly DFT simulations while maintaining the prediction accuracy at the DFT level. Specifically, we translate training molecules into text-based identifiers- SMILES. Following that, we parametrize training matrices with sets of short-range descriptors coupled with long-ranged descriptors. Finally, we use linear and Gaussian process regressions (GPR) to regress over the linear and nonlinear matrix systems. To our knowledge, this is the first work that encompasses molecular parsing at the first and second nearest neighbors throughout the featurization, training, and deployment stages. We trained and validated our models with more than 400 surface species on Pt, Ru, and Ir surfaces with diverse surface species. Results exhibit robust performance to reproduce the energies of interest. Notably, predictions take seconds to conclude, and the prediction accuracy is on par with actual DFT, as the R2 parity score benches nearly to perfect 1, and the mean absolute error (MAE) is as low as 0.04 eV.
Finally, we will present how we can navigate through complicated reaction networks and demystify reaction mechanisms by ascertaining the changes in thermodynamical quantities, for example, Gibbs free energy at various temperatures on modeled metal catalysts.