2024 AIChE Annual Meeting
(511c) Computational Modelling of Dynamic Charge and Adsorption Responses
Authors
Cristina Pizzolitto, CASALE SA
Pierdomenico Biasi, CASALE SA
Paul J. Dauenhauer, University of Minnesota
Turan Birol, University of Minnesota
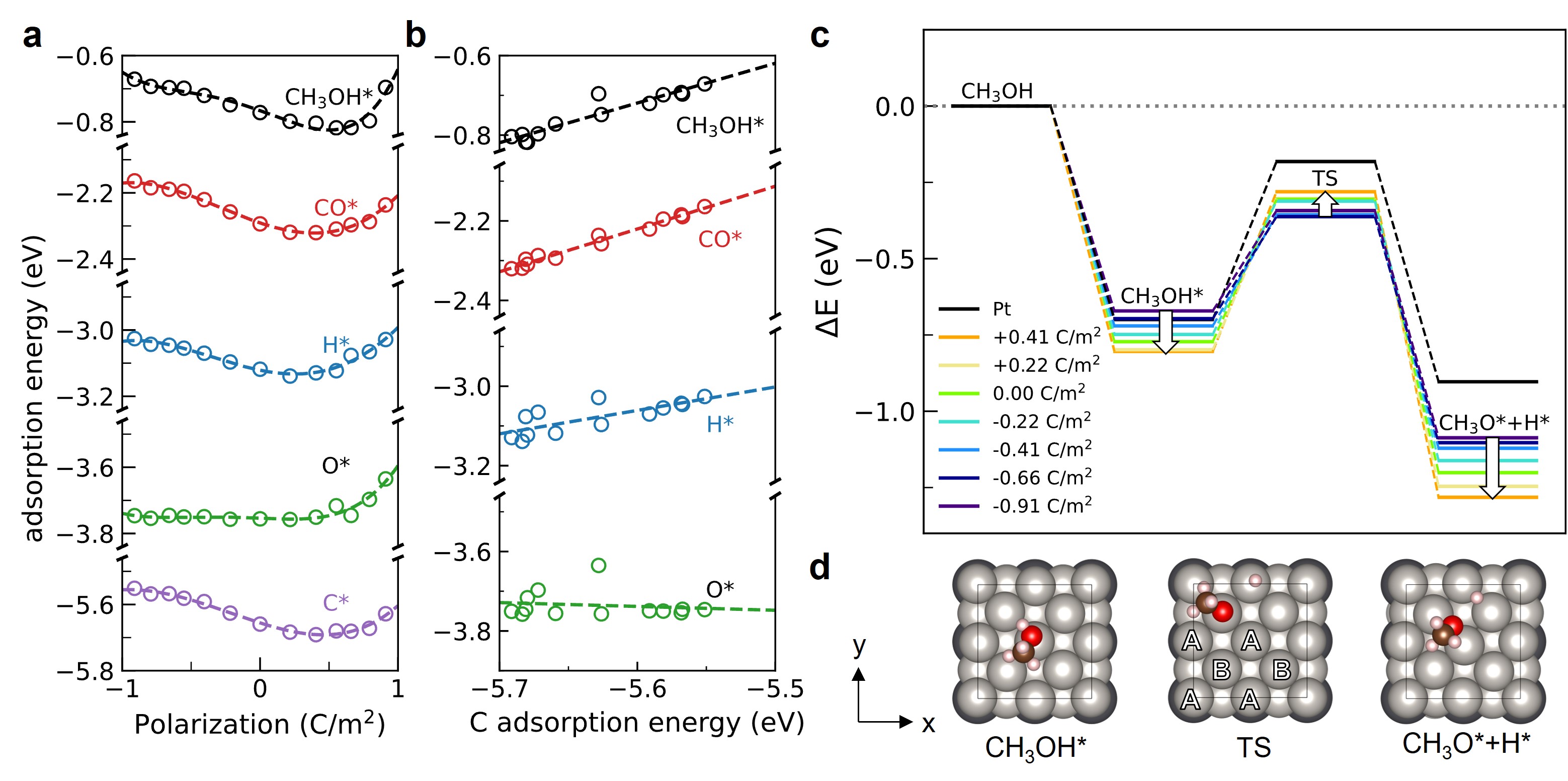
The Sabatier principle and the scaling relations have been widely used to search for and screen new catalysts in the field of catalysis. However, these powerful tools can also serve as limitations of catalyst control and breakthrough. Dynamic programmable catalysis, where surfaces oscillate between two or more states with distinct binding energies of key reaction intermediates, is a promising strategy to overcome the limitations imposed by the Sabatier principle. In this talk, we propose efficient methods to computational model the dynamically perturbed states of catalysts from first principles, such as using electric potential to polarize the support. The results allow us to study charge and adsorption of catalysts by external modulation. First, we demonstrate that the properties of catalysts are determined by support polarization, irrespective of the magnitude of spontaneous polarization of support. The approach enables elucidating the scaling relations between binding energies at various polarization values of support. Moreover, we observe the breakdown of scaling relations for the surface controlled by support polarization. By studying the surface electronic structure and decomposing the induced charge into contributions from different atoms and orbitals, we identify the inherent structural property of the interface that leads to the breaking of the scaling relations. Specifically, the displacements of the underlying oxide support impose its symmetry on the catalyst, causing the scaling relations between different adsorption sites to break.
Figure caption: (a) Changes of adsorption energies on top site 2-layer Pt(100) for CH3OH*, CO*, H*, O*, and C* with varying polarization of underlying support PbTiO3. (b) The adsorption energies scale linearly with the adsorption of carbon atoms. (c) Reaction coordinate of O-H bond dissociation of methanol at Pt(100) surface, on various polarized PbTiO3 support. (d) Reaction scheme of methanol dissociation.