2024 AIChE Annual Meeting
(4bp) Multiscale Modeling and Analysis of Adsorption Based Processes: Applications to Hydroisomerization of Alkanes and Breakthrough Curve Modeling
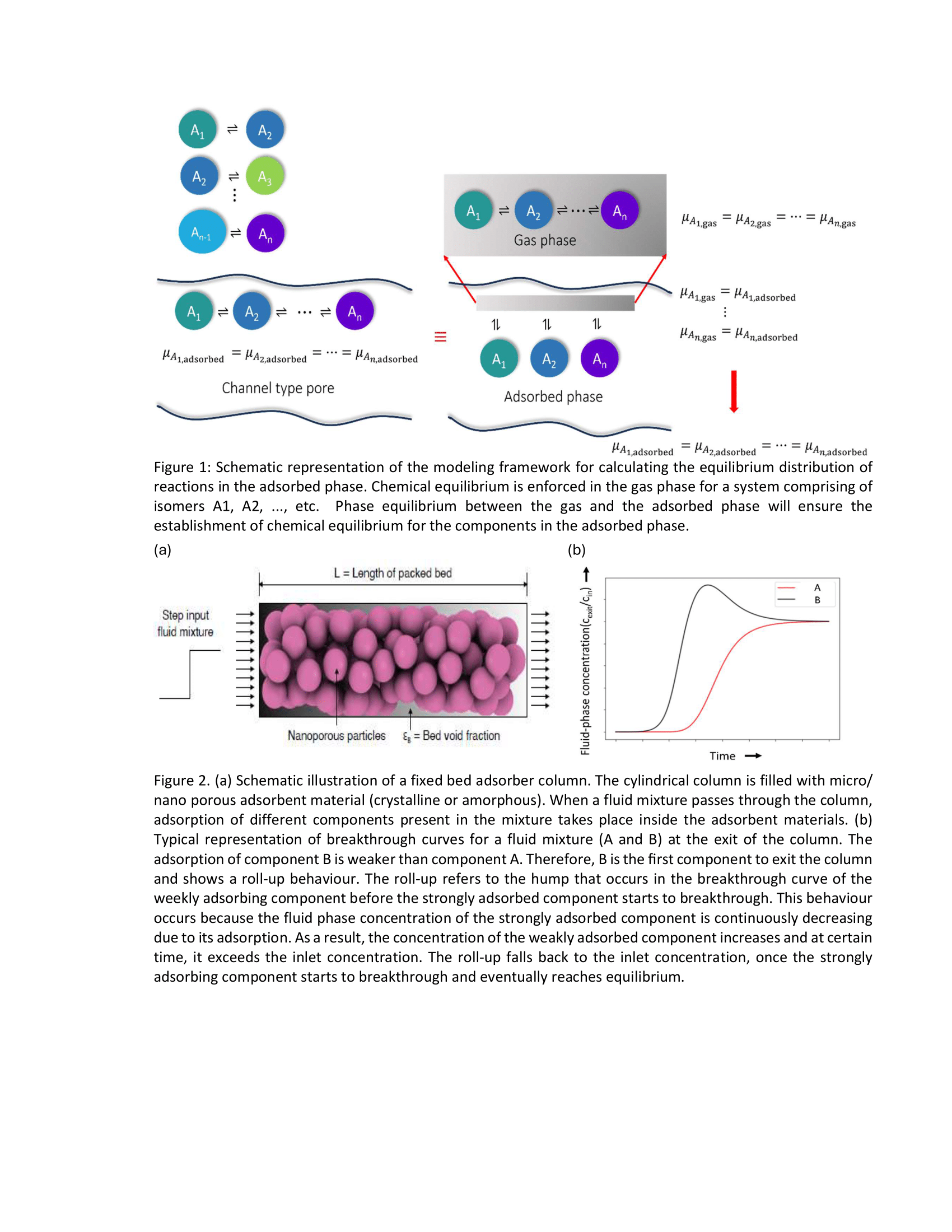
I have also developed an open-source software called RUPTURA [2] in collaboration with my PhD supervisors Prof. dr. ir. Thijs J. H. Vlugt and Dr. David Dubbeldam and other collaborators. This software simulates breakthrough curves, predicts mixture adsorption isotherms, and performs fitting of pure component adsorption isotherms. Estimation of adsorption isotherms are crucial in designing adsorption-based processes which quantify the affinity between the adsorbate molecules and the adsorbent material. Many industrial separation processes take place at dynamic conditions where, the concentration of the fluid phase components measured at the outlet of the adsorption column as a function of time are known as breakthrough curves (Fig. 2). Both adsorption isotherms and breakthrough curves can be obtained experimentally but this process is very time consuming as many experiments are required to generate these datasets. Therefore, one can always opt for modeling and simulation-based estimation of these curves. Acknowledging the importance of the adsorption isotherms and breakthrough curves calls for the need of an open-source software which can compute these curves. Hereby, we present RUPTURA code (https://github.com/iraspa/ruptura) as a free and open-source software package (MIT license) [2]. RUPTURA is capable of (1) simulating breakthrough curves for adsorption of gas phase molecules at isothermal conditions, (2) predicting mixture isotherms using methods like the Ideal Adsorption Solution Theory (IAST), Segregated-IAST (SIAST), and explicit isotherm models [3], and (3) fitting of pure-component isotherm models on computed or experimentally measured pure component adsorption isotherm data. For breakthrough curve simulations, RUPTURA supports both step and pulse style input for the feed mixture. The code supports a wide variety of isotherm models like Langmuir, Anti-Langmuir, BET, Henry, Freundlich, Sips, Langmuir-Freundlich, Redlich-Peterson, Toth, Unilan, O'Brian & Myers, Asymptotic Temkin, and Bingel & Walton. Mixture prediction and breakthrough simulations rely on these isotherm models that are obtained by fitting pure component isotherm data. In RUPTURA, fitting of pure-component isotherms are performed using genetic algorithm. Breakthrough plots and animations depicting the variation of properties like fluid velocity, column pressure, and adsorbed loadings along the fixed bed are automatically generated. We foresee our code being used in (industrial) research for screening adsorbents for separation processes based on Pressure Swing Adsorption (PSA), and Temperature Swing Adsorption (TSA) but also for teaching in chemistry and chemical engineering classes.
Research Interests
My research interest lies in the study of various chemical, electrochemical, and thermal engineering processes through multiscale modeling and simulations. This includes numerical modeling, molecular simulations, and computational fluid dynamics (CFD). Apart from adsorption based systems, I have also worked on other applications in the field of sustainable process and energy systems. This includes understanding the role of electrochemical promotion of catalysis on reaction product distribution of Fischer Tropsch reaction, numerical modeling of fuel assisted solid oxide electrolysis, and study of melting phenomenon of encapsulated phase change materials in packed bed thermal energy storage systems.
Acknowledgements
I am thankful to my collaborators Dr. Marcello S. Rigutto, Dr. Erik Zuidema, Dr. Richard Baur, and Dr. Umang Agarwal from Shell Global Solutions International B.V., Amsterdam, The Netherlands, Dr. Salvador R. G. Balestra from Department of Physical, Chemical and Natural Systems, Universidad Pablo de Olavide, Sevilla, Spain, Prof. dr. Sofia Calero from Eindhoven University of Technology, Eindhoven, The Netherlands, Dr. Silvia Lasala from Université de Lorraine, Nancy, France, Josh J. Sleijfer, Jeroen Op de Beek, Stach van der Zeeuw, and Daniil Zorzos from Delft University of Technology, Delft, The Netherlands. This work was sponsored by NWO Domain Science for the use of supercomputer facilities. This work is part of the Advanced Research Center for Chemical Building Blocks, ARC-CBBC, which is co-funded and co-financed by the Netherlands Organization for Scientific Research (NWO) and the Netherlands Ministry of Economic Affairs and Climate Policy.
References:
[1] S. Sharma, M. S. Rigutto, E. Zuidema, U. Agarwal, R. Baur, D. Dubbeldam, and T. J.H. Vlugt. “Understanding shape selectivity effects of hydroisomerization using a reaction equilibrium model”, Journal of Chemical Physics 160, 214708 (2024).
[2] S. Sharma, S. R.G. Balestra, R. Baur, U. Agarwal, E. Zuidema, M. S. Rigutto, S. Calero, T. J. H. Vlugt, and D. Dubbeldam. "RUPTURA: simulation code for breakthrough, ideal adsorption solution theory computations, and fitting of isotherm models." Molecular Simulation 49, 893-953 (2023).
[3] S. Sharma, M. S. Rigutto, R. Baur, U. Agarwal, E. Zuidema, S. R.G. Balestra, S. Calero, D. Dubbeldam, and T. J.H. Vlugt. "Modelling of adsorbate-size dependent explicit isotherms using a segregated approach to account for surface heterogeneities." Molecular Physics 121, e2183721 (2023).