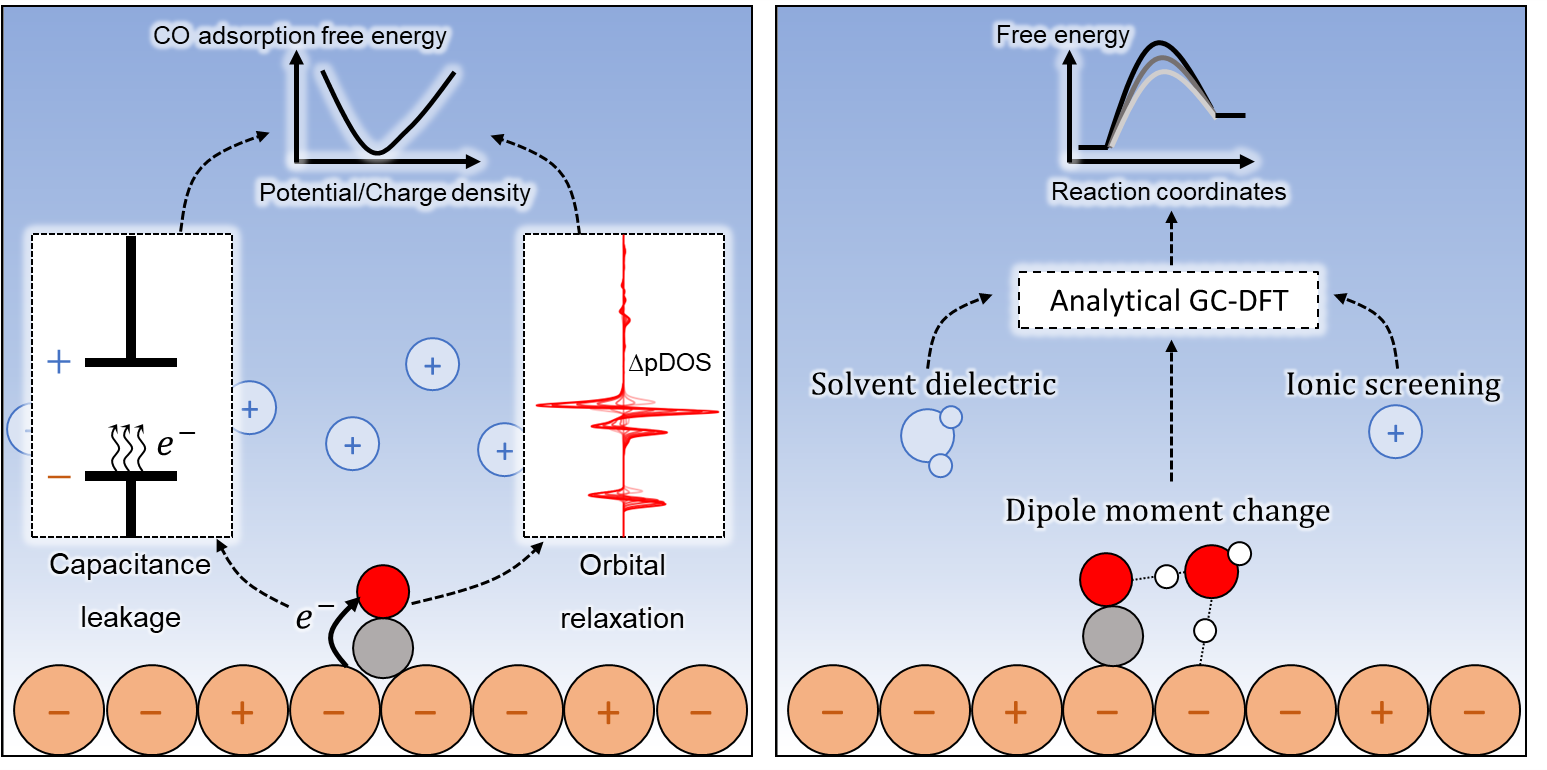
CO adsorption/desorption and surface reactions on electrodes are ubiquitous elementary steps in electrocatalytic CO
2 reduction or C-N coupling reactions. These elementary steps involving adsorbed *CO are conventionally treated as chemical steps, i.e., with no charge transfer and therefore, no dependence on the electrode potential. By employing a grand-canonical DFT approach, we demonstrate and explain the potential-dependent energetics for *CO adsorption and surface reactions on Cu electrodes. On Cu(111) and Cu(100), the adsorption free energy of CO varies nonlinearly with the applied potential, increasing by up to 0.15 eV across the working potential range of CO
2RR (0.0 to -1.0 V vs. SHE) and exhibiting a minimum at around -1.0 V. Electronic structure analysis reveals a connection between the nonlinearity of potential-dependent CO adsorption free energy and a local charge transfer between surface Cu atoms and *CO, which manifests into an interfacial capacitance loss. CO adsorption becomes weaker below -1.0 V which is attributed to *CO orbital relaxation and an increased Pauli repulsion. The activation free energy for three *CO surface reactions (forming *COH, *CHO, and *OCCO) varies with potential at different degrees depending on the change in surface-normal dipole moment across the reaction coordinate. By compartmentalizing the grand free energy using an analytical approach, the potential dependence is further rationalized by the physical interpretation of the electrolyte model parameters such as the solvent dielectric constant and the ionic screening length. Overall, these findings contribute fundamental insights into the potential-dependence for conventionally categorized chemical steps in electrocatalysis.