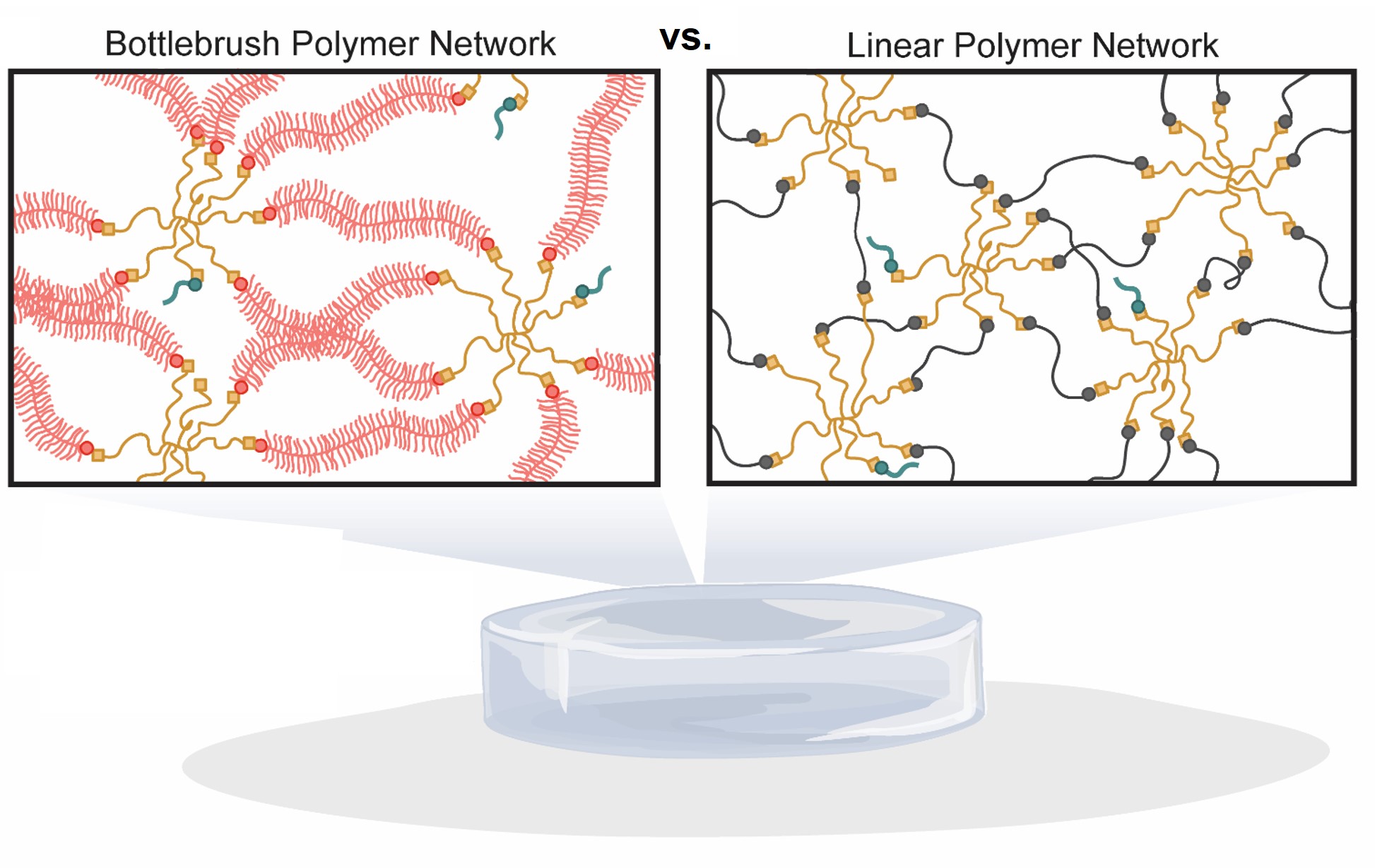
Bottlebrush polymers are composed of polymer chains densely grafted to a linear polymer backbone. These side chains extend radially to fill space causing the linear polymer backbone to extend and become rigid. Due to the space filling nature of the polymer chains surrounding the polymer backbone, bottlebrush polymers behave as flexible rods in solution and have high (> 100 kDa) molecular weights of entanglement. This extended conformation presents a unique building block with which to engineer step growth polymer networks connected by non-entangled polymer chains leading to greater network diffusivity, decreased bulk moduli, and strain-stiffening mechanical properties. The poly(polyethylene glycol) monomethyl ether acrylate (PPEGA9) dithiol bottlebrush polymers used in this work were synthesized using a di-functional trithiocarbonate chain transfer agent via reversible addition fragmentation chain transfer polymerization techniques to yield four PPEGA9 with increasing backbone degree of polymerization (
Nbb = 20, 50, 80, and 110). The trithiocarbonate end-groups were cleaved to free thiols using aminolysis before each PPEGA9-
Nbb dithiol was crosslinked into a hydrogel network with an 8-arm, 20 kDa PEG-norbornene using 365 nm light in the presence of a water-soluble photoinitator. The evolution of network formation and nonlinear elastic mechanical properties of each bottlebrush polymer hydrogel were characterized
in situ using oscillatory shear rheology. Keeping the initial polymer gel fraction the same in each formulation, the gelation time and in situ plateau moduli were observed to decrease with increasing backbone degrees of polymerization. Once swollen, the hydrogels were measured to have shear moduli ranging from 300 Pa to 12 Pa, with the PPEGA9-110 having the lowest modulus. These ultrasoft networks also exhibited nonlinear elastic behavior as a function of applied stress. Each hydrogel formulation stiffened at a designated critical stress (75 to 2 Pa) that decreased as the length of the PPEGA9-
Nbb macromer increased. Due to the rod-like, extended morphology of the bottlebrush polymer macromers, only a small amount of applied stress was necessary to sufficiently deform the network to fully extend the polymer chains connecting the covalently crosslinked network. To investigate how the bottlebrush polymer length changed the pore size of the ultrasoft networks, the diffusion coefficient of fluorescently labeled bovine serum albumin was measured within the network using fluorescence recovery after photobleaching techniques. The diffusion coefficient increased as a function of increasing backbone degrees of polymerization. Therefore, this suggests that the bottlebrush polymer macromers act as rigid struts connecting the crosslinked network resulting larger pore sizes which allow for faster diffusion. In this talk we will investigate how bottlebrush polymer degree of polymerization can be used to tune the mechanical properties of the resulting hydrogels and their potential as platforms to study how cells respond to ultrasoft and strain-stiffening microenvironments.