2023 AIChE Annual Meeting
(676d) Catalytic Trends for CO2 Hydrogenation Reactivity and Selectivity on Transition Metals
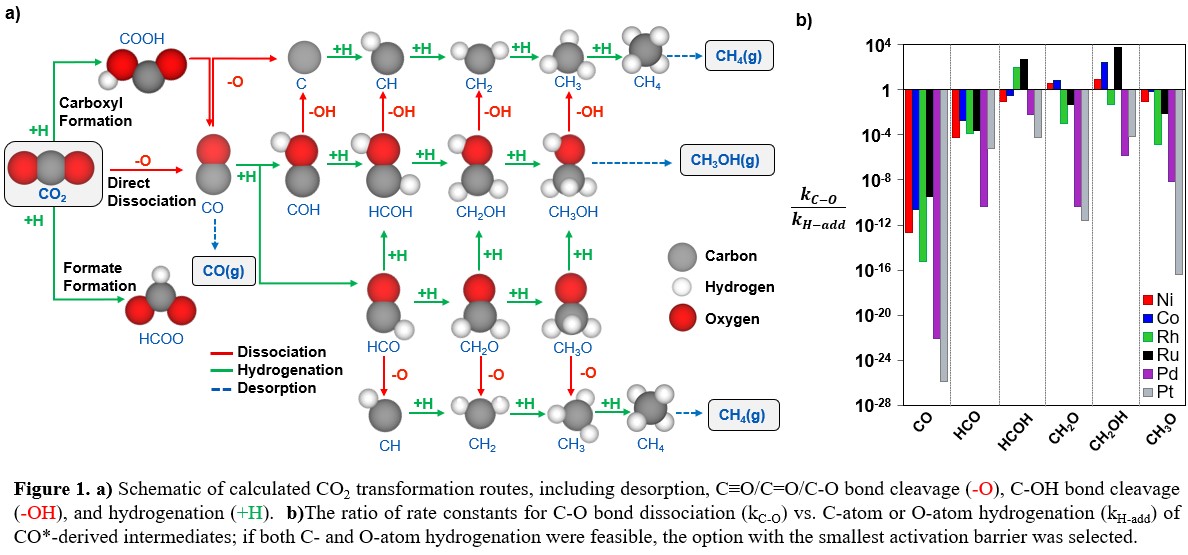
CO2 hydrogenation catalyzed by transition metals can yield C1 products (e.g., CH4, CH3OH, CO) at relatively mild conditions (473-673 K, 1-10 bar). CH3OH and CH4 often form in parallel with CO, motivating questions about catalytic structure-function relationships that drive reactivity and selectivity.1,2 This work provides mechanistic insights into CO2 hydrogenation routes (Fig. 1a), through a systematic comparison of the activity of low-index surfaces (Co, Ni, Pd, Pt, Rh, and Ru), using density functional theory (DFT; PBE-D3) calculations and microkinetic models. DFT results indicate that oxophilic transition metals (Co, Ni, Rh, and Ru) activate CO2 via direct dissociation, while less oxophilic metals (Pd and Pt) require hydrogen assistance. Selectivity between CO, CH4, and CH3OH is driven by preferences for CO desorption, hydrogenation, and C-O bond dissociation. Two C-O bond dissociation steps are required to form CH4, whereas only one is required for CH3OH or CO. The kinetic preference for dissociation v. hydrogenation is reflected in rate constant ratios (kC-O:kH-add) for CO*-derived species (Fig. 1b). C-O bond dissociation is kinetically challenging on Pd and Pt, as indicated by kC-O:kH-add < 1, but favorable on Co, Ni, Rh, and Ru after two hydrogenations of CO*. Microkinetic modelling indicates that CH4 forms via a kinetically relevant C-O bond dissociation step on Co, Ni, Rh, and Ru; Pd and Pt favor CO production, through carboxylate dissociation and CO* desorption. The comprehensive data set offered herein permits facile comparisons across transition-metal surfaces for C1 reactions, providing insight on the fundamental nature of their active sites and key steps that control product selectivity. Ultimately, the results from this study will advance design strategies for novel materials used in C1 chemistries, such as carbon capture or utilization processes.
[1] J. Am. Chem. Soc. 2017, 139, 29, 9739â9754
[2] ACS Catal. 2020, 10, 19, 11318â11345