2023 AIChE Annual Meeting
(650b) First Principles Study of Copper Cathodic Corrosion during CO2rr
Authors
Hori Pada Sarker - Presenter, SUNCAT Center for Interface Science and Catalysis, Department of Chemical Engineering, Stanford University, 443 Via Ortega
Frank Abild-Pedersen, SLAC National Accelerator Laboratory
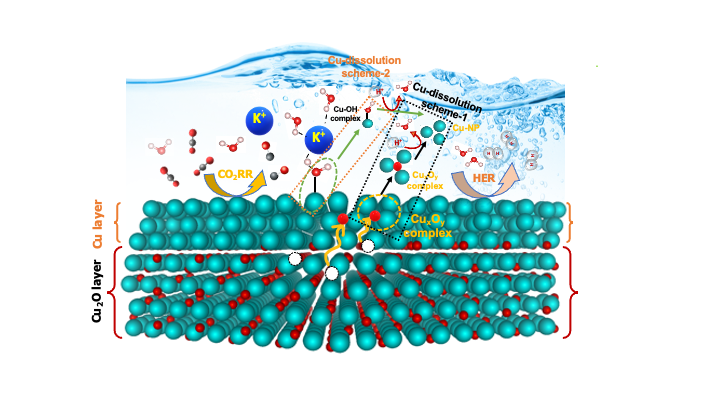
Electrochemical CO2 reduction reaction (CO2RR) offers an efficient strategy to upcycle anthropogenic CO2 gas into value-added fuels. Until now, copper (Cu) is the first choice as a catalyst for electrochemical CO2 reduction. While Cu-based electrocatalysts can form desirable C2+ products, they have shown drastic change in structure and morphology which eventually limits its stability. During CO2RR, surface Cu-atoms go through cathodic corrosion, and as a result, leach out from surface to electrolyte via intermediate Cu-compounds which later reduce back to elemental Cu and form nanoparticle either in solution or redeposit to surface. However, Cu dissolution mechanism during CO2RR is still a highly debatable issue within CO2RR community. In this study, we aim to understand the detail thermodynamics and kinetics of Cu dissolution mechanism during CO2RR to design stable Cu-catalyst. Herein, a first principles density functional theory (DFT) approach is used to simulate Cu-corrosion under CO2RR conditions. As a model surface, we employed Cu (211) surface with a kink site. We assume that oxygen atoms from the oxide bulk diffuse to Cu-surface and form transient CuxOy intermediate which eventually get dissolved and later reduced back to elemental Cu-atoms. The thermodynamic stability of such CuxOy intermediate compounds were tested in terms of Gibbs free energy derived from DFT calculations. In addition to thermodynamic stability, we also studied the kinetics in terms of diffusion barrier calculations. We implemented nudged elastic band (NEB) method to estimate the diffusion barriers. We also investigated the role of applied potential, pH, internal strain due to the lattice mismatch as well as the reaction intermediate species i.e., hydrogen (H), hydroxyl ion (OH-) etc., towards the Cu corrosion process. The data obtained from theoretical calculations were also corroborated with available experimental observations. Finally, the details of this study will be presented during the conference.