2023 AIChE Annual Meeting
(362i) Advancing Catalyst Design through Insights from Computational Modeling of Heterogeneous Catalysis
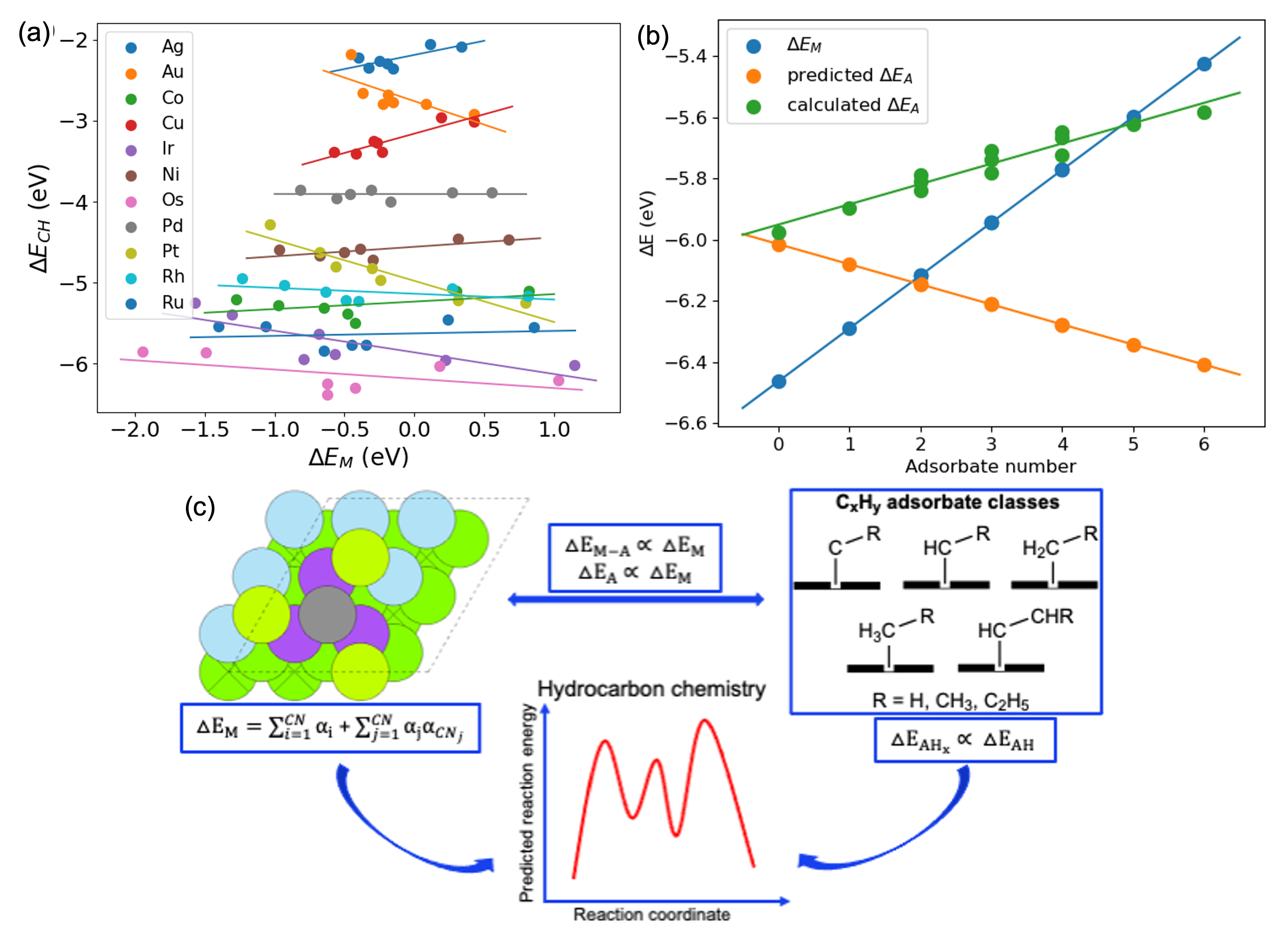
One way to accelerate the design of heterogeneous catalysts is to identify relevant descriptors that accurately link binding and activation energies to reactivity. We investigated scaling relations between binding energies of various hydrocarbon-based adsorbates on three different Pt surfaces and metal binding energies estimated via the recently developed coordination-based α-scheme model. The coordination-based α-scheme model can predict metal binding energies very accurately and efficiently. The model predicted metal binding energies that showed a strong correlation with the metal adsorbate and adsorbate binding energies (ÎEPt-A and ÎEA). We found that scaling slopes are similar for certain groups of adsorbates, thus enabling a classification of adsorbates based on their spatial and electronic structure, providing fast description of binding strengths for each member of the class. In case of hydrocarbon chemistry, various classes of adsorbates are linearly correlated with CH and can serve as a descriptor for dehydrogenation of hydrocarbons. To understand the relationships between different metals, we investigated a range of metals, including Ag, Au, Co, Cu, Ir, Ni, Os, Pd, Pt, Rh, and Ru. At a specific coordination environment, the binding energies of CH can be determined for other metals from any metals considered. All these correlations are based on the coordination-based α-scheme model, making it easier to understand the complexities of hydrocarbon chemistry on different metals and obtain reaction energies without extensive computational calculations (Figure 1a and 1c).
In heterogeneous catalysis, the interaction among adsorbates can strongly influence reaction rates, making it essential for understanding intrinsic kinetics and catalytic activity. The proposed model considers the modification of the metal surface caused by the presence of other adsorbates in the surrounding. The binding energy of metal atoms for the same type of adsorbates decreases as the number of C, H, or O adsorbates in their surroundings on the Pt(111) surface increases. It is important to note that the presence of other adsorbates can significantly affect the adsorption behavior of a specific adsorbate, and these effects cannot be explained solely by the electronic structure. The electrostatic interactions among various adsorbates also play a crucial role. The changes in predicted and calculated adsorption energies of C, H, and O on the Pt(111) surface in the presence of other adsorbates exhibit a linear scaling behavior with the number of adsorbates present in the surroundings, indicating that the electrostatic interactions among various adsorbates depend on the type and number of adsorbates. By scaling with the number of adsorbates, the electronic and electrostatic effects can be determined, and the metal binding energies can be utilized to predict the adsorbate binding energies. Taking into account the electronic and electrostatic components and the presence of different types of adsorbates, the model offers a significant advantage in predicting the strength of adsorbate-adsorbate interaction (Figure 1b).
These approaches can be extended to investigate other complex reactions. This can be achieved without the need for extensive computational calculations, making it a valuable tool for both theoreticians and experimentalists in the field of heterogeneous catalysis. Hence, this suggests a generalizable scheme for designing active catalyst for various complex reactions rapidly and accurately. While adsorbate-adsorbate interactions and correlations are crucial steps toward understanding heterogeneous catalysis, it is only part of the puzzle. To achieve the ultimate goal of designing and synthesizing highly active and selective catalysts, other important factors must also be taken into account. For example, the kinetic properties of the reaction must be considered, including the activation energy and rate-limiting steps. Additionally, the effect of alloying, or the addition of other metals to the catalyst, can have a significant impact on its performance. Ultimately, the most important test of a catalyst's activity and selectivity is experimental synthesis and testing. The predictions made through computational models and simulations must be verified through real-world experiments. This requires the development of reliable synthetic methods for creating the predicted active catalyst and testing its reactivity under realistic reaction conditions.
Figure 1: (a) The α-scheme model predicts metal binding energies, which scales with the CH adsorbate binding energies. (b) The metal binding energy can be used to predict the adsorbate binding energies. By considering changes in the electronic structure the calculated adsorbate binding energy can be plotted as a function of the number of adsorbates present. (c) The correlation between metal adsorbate and adsorbate binding energies with metal binding energies can be analyzed, and hydrocarbons can be classified. The α-scheme model predicted metal binding energies, and linear scaling relations with metal adsorbate and adsorbate binding energies can be used to predict reaction energies for the dehydrogenation of hydrocarbons.