2022 Annual Meeting
(176a) Dynamically Formed Active Sites on Liquid Boron Oxide for Selective Oxidative Dehydrogenation of Propane
Authors
Gregory Collinge - Presenter, Pacific Northwest National Laboratory
Jinshu Tian, Pacific Northwest National Laboratory
Simuck Yuk, United States Military Academy
Jingdong Lin, Xiamen University
Wei Shou, Zhejiang University of Technology
Vassiliki-Alexandra Glezakou, Pacific Northwest National Laboratory
Mal-Soon Lee, Pacific Northwest National Laboratory
Yong Wang, Washington State University
Roger Rousseau, ORNL
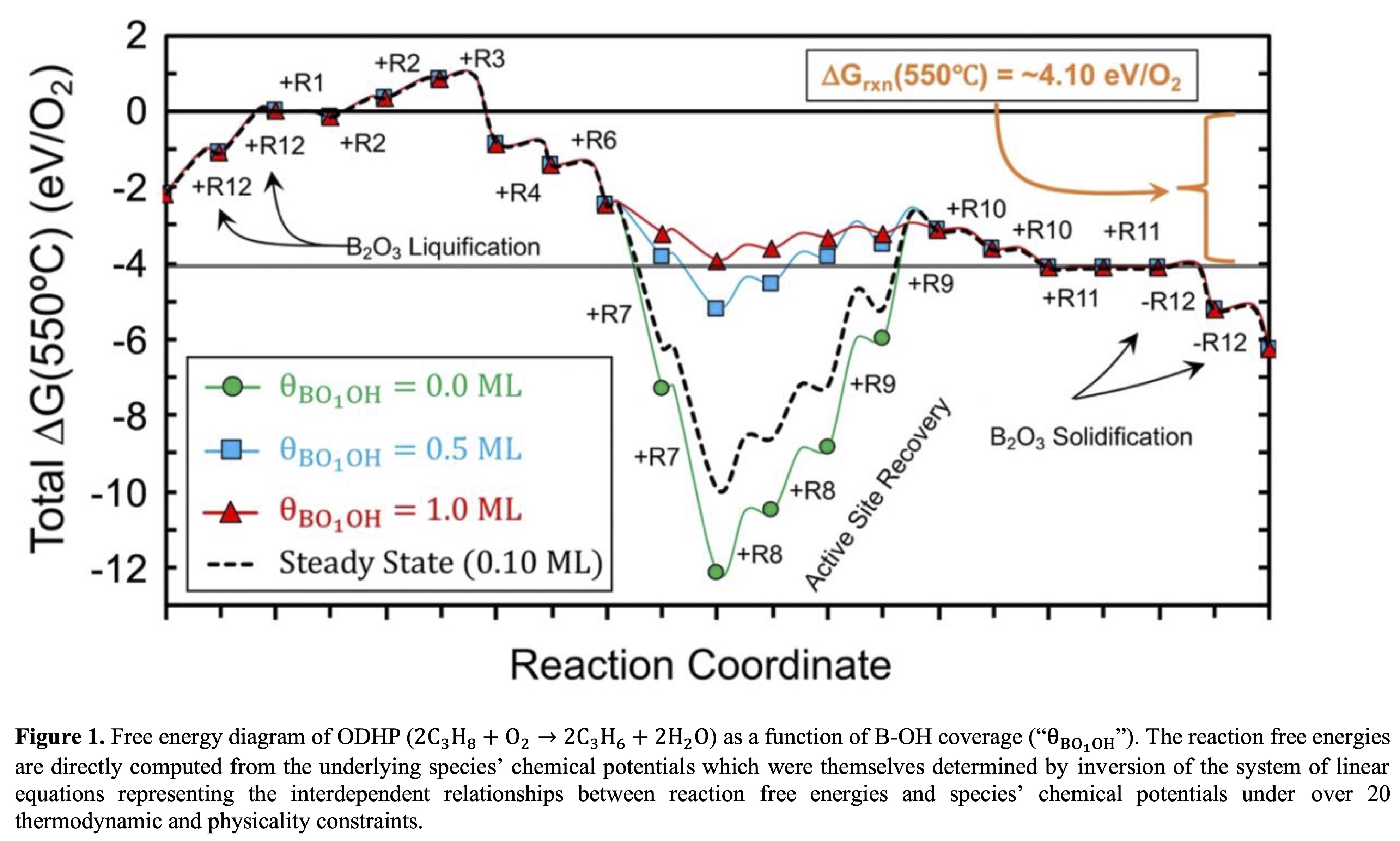
Boron-oxide-based catalysts have been shown to be both active and selective for driving the oxidative dehydrogenation of propane (ODHP) without the use of metal promoters. However, this reaction typically occurs at temperatures where boron oxide melts, challenging experimental identification of the molecular structures within the boron oxide phase under reaction conditions and thus hindering understanding of its active sites and reaction mechanism(s). By combining DFT calculations, ab initio molecular dynamics simulations, in situ Raman characterization, and thermodynamically self-consistent microkinetic modeling, we propose that the di-coordinated boron sites (>B*) that are dynamically formed in liquid boron oxide are the active species for O2 activation under reaction conditions. This peroxy-like species (>B-O-O-B<) can be viewed as a moderate oxidant for ODHP, reactive to propane but inert to propene. The dynamical >B-O* dangling bonds, originating from the >B-O-O-B< site as well as the liquid B2O3 structure itself, play a critical role in the abstraction of H atoms from propane (adsorbed C3H7* radical formation). In fact, microkinetic modeling reveals that the formation of adsorbed C3H7* radicals is the main rate controlling step (~75% rate control) due to highly endergonic adsorption of propane into the system. Recovery of the di-coordinated >B* active sites then controls the remainder of the rate (~25% rate control) due to a strong dependence of water formation and desorption chemical potentials and activation barriers on surface B-OH concentration (Figure 1), which we find must reach saturation for the barriers to become surmountable. These findings provide significant insights into the active sites and reaction mechanisms of ODHP on boron-based catalysts and emphasize the importance of understanding and accurately modeling the liquid nature of the catalyst to account for its catalytic activity.