2022 Annual Meeting
(173d) Multiscale Modeling Approach for Designing Novel Hierarchical Carbon Cathodes for Ultrahigh Capacity Aprotic Li-O2 Battery
Owing to current challenges and limitations, improving the discharge capacity of Li-O2 battery (LOB) have become critically acclaimed topic among the scientific community. Using experimental studies [2], particular attention has been paid to the optimization and designing of novel cathode materials to overcome the challenges of discharge product deposition. However, multiphysics/multiscale processes occurring in LOB are very difficult to be captured by experimental approaches. Therefore, utilization of a multiscale modeling technique is indispensable to capture various phenomena happening at varying time and length scale, from molecule to system level [3]. The multiscale modeling approach is spanned over the continuum models at macroscopic level to classical forcefield models, reactive forcefield approaches, and the fundamental quantum chemical models (DFT, and ab-initio MD) at the microscopic level. Quantum studies (such as DFT and AIMD) are useful techniques to investigate the reaction-diffusion mechanism, and thermodynamic characteristics during the Li-O2 battery operation [4]. Unfortunately, such atomistic dynamic studies are restricted to small length scales (~ 100 atoms) and time scales (~ fs to ps). Alternatively, classical forcefield are quite inexpensive computationally and used to model systems at larger time and length scales. However, they do not consider bond breakage or formation during molecular interactions, and hence cannot be employed for reactive systems. Reactive forcefield (reaxFF), firstly developed by van Duin [5], is a bond order based forcefield which has potential to describe the chemical bonds formation and breakage. ReaxFF, like classical forcefields, also incorporates the van der Waals and Coulombic interactions among the molecules. Therefore, ReaxFF are being used to bridge the gap between quantum studies and classical forcefield methods.
In this work, we have developed multiscale top-down approach, in which we started from continuum modeling of Li-O2 battery cell [6]. We propose different hierarchical cathode designs with distributed tortuosity, and distributed porosity which led to improved discharge capacity by approximately 50 %. It shows that changing pore size and volume, and pore alignment has higher impact on the species (Li, O2) transport and solid discharge product (Li2O2) formation leading to varying LOB performance. Further, we have observed that Li2O2 deposition mechanisms (i.e., surface passivation and solution growth) not only depends on the cathode architecture but also the discharge current density. Higher discharge current density leads to film like deposition of Li2O2 on the cathode which induces surface passivation. On contrary, lower discharge current density promotes the growth of Li2O2 in large toroidal form leading to pore clogging. Considering the continuum modeling findings, it is imperative to dig deep and uncover the impact of structural properties on the electrochemical kinetics, Li2O2 deposition and distribution, and species diffusion barriers. For this, we moved one step down towards reactive forcefield based molecular modeling (reaxMD) study of the cathode structural properties and their impact on battery performance.
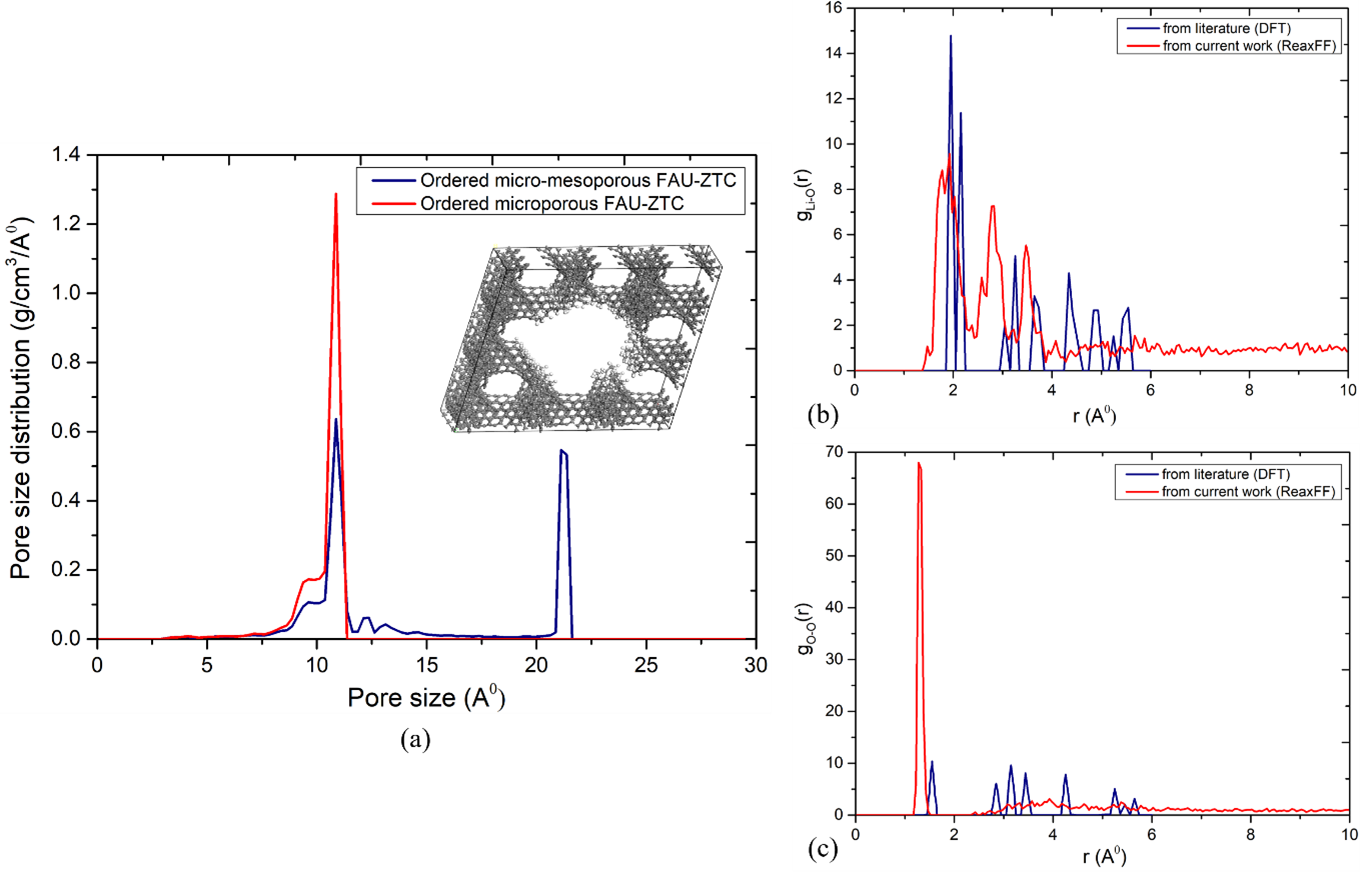
In our study, we have employed zeolite templated carbons (ZTCs) for LOB cathodes owing to their ultrahigh surface area, and ordered micro-porous framework. In previous work, ZTCs have been employed as secondary battery electrode [7] due to their relatively high conductivity and tunability of structure, however, their potential for LOB cathode never explored. We have performed reactive molecular dynamics simulations of different structures of ZTC such as FAU-ZTC and MFI-ZTC to exploit the diffusion coefficients of the species (including DMSO, LiPF6, O2, and Li2O2), discharge reaction pathways, and deposition of Li2O2. The single unit cells were taken from ZTC materials cloud database [8]. Moreover, the effect of hierarchical micro-mesopores was evaluated. The resulted structures (Figure 1, a) lead to enhanced specific surface area as well as pore volume. For the obtained Li2O2 system, the basic structural characteristics were studied by pair distribution functions for Li-O and O-O (calculated by reaxFF) and compared with the DFT study [9]. The position of the first peaks in both graphs (Figure 1, b and c) shows the Li-O (i.e., 1.925 Å) and O-O (i.e., 1.275 Å) neighbor distances within the first coordination shell. Compared to Li-O pair, the narrow distribution of O-O pair in first coordination shell in Li2O2 structure is because of the presence of unique O-O covalent bond of O2-2. This narrow pair distribution gives the O-O bond length of Li2O2 within the range of 1.275-1.375 Å. On contrary, the broader pair distribution of Li-O is attributed to the coupling of Li+ with various O2-2. orientations leading to the Li-O bond distances within the range of 1.775-2.875 Å. Furthermore, the self-diffusivity results of Li-ion and oxygen reveals that, prior to discharge process, both species move randomly through the tunnels of FAU-ZTC which results in quite similar diffusivities i.e., 4.5E-09 for oxygen and 5E-09 for Li-ions along x-direction. However, as discharge process continues and Li2O2 forms, the movement of both Li-ion and oxygen become restricted due to the formation of chemical bonds between Li-ion and oxygen. this effect can be further refined by running the simulation for longer time duration.
In conclusion, from our multiscale simulation study, we have deduced that O-O bond length might have impact on the crystalline structure of Li2O2, which restricts species transport upon high growth. Therefore, ordered micro-mesoporous FAU-ZTC can provide enough space to store large amount of Li2O2 without restricting the flow of ions and hence prolong the discharge process. This highlights the need for multiscale modeling approach that guides the design and selection of materials with enhanced electrochemical properties in terms of energy density.
Fig. 1: Schematics of (a) pore size distribution in FAU-ZTC, and pair distribution of (b) Li-O and (c) O-O inside Li2O2. Here DFT studies are taken from [9]
References
[1] Z. Ma, X. Yuan, L. Li, Z.F. Ma, D.P. Wilkinson, L. Zhang, J. Zhang, Energy Environ. Sci. 8 (2015) 2144â2198.
[2] J.W. Jung, S.H. Cho, J.S. Nam, I.D. Kim, Energy Storage Mater. 24 (2020) 512â528.
[3] K. Hayat, L.F. Vega, A. AlHajaj, Renew. Sustain. Energy Rev. 154 (2022) 111849.
[4] X. Luo, R. Amine, K.C. Lau, J. Lu, C. Zhan, L.A. Curtiss, S. Al Hallaj, B.P. Chaplin, K. Amine, Nano Res. 10 (2017) 4327â4336.
[5] M.M. Islam, V.S. Bryantsev, A.C.T. Van Duin, J. Electrochem. Soc. 161 (2014) E3009.
[6] K. Hayat, L.F. Vega, A. AlHajaj, J. Electrochem. Soc. 168 (2021) 120534.
[7] J. Miao, Z. Lang, T. Xue, Y. Li, Y. Li, J. Cheng, H. Zhang, Z. Tang, Adv. Sci. 7 (2020) 2001335.
[8] E.E. Taylor, K. Garman, N.P. Stadie, Chem. Mater. 32 (2020) 2742â2752.
[9] K.C. Lau, D. Qiu, X. Luo, J. Greeley, L.A. Curtiss, J. Lu, K. Amine, Energies. 8 (2015) 529â548.